Bem-vindo ao mundo de Marvin! 
Este manual foi criado para ajudar os usuários a utilizar o HPCC Marvin. Apesar de ser um pouco mal-humorado às vezes, Marvin está sempre disposto a te ajudar!
"Você acha que VOCÊ tem problemas? Experimente ser um robô maníaco depressivo..."
— Marvin, O Guia do Mochileiro das Galáxias
O que é HPCC?
HPCC (High Performance Computing Cluster) é um sistema de computação que combina múltiplos computadores (chamados nós) para trabalhar juntos em tarefas complexas, que requerem grande poder de processamento. O HPCC é projetado para lidar com grandes volumes de dados e realizar cálculos intensivos, tornando-o ideal para aplicações científicas, simulações, modelagem e análise de dados.
Sobre o HPCC Marvin
Marvin é nome do HPCC do LNBio/CNPEM, adquirido em 2022 da empresa Atos, empresa especializada em soluções de HPC e responsável por alguns dos maiores supercomputadores do Brasil e do mundo, como o Santos Dumont.
O HPCC Marvin está hospedado no Data Center do Sirius/LNLS. Para informações sobre a arquitetura do HPCC Marvin, veja a seção Arquitetura.
Acesso ao HPCC Marvin
Para começar a utilizar o HPCC Marvin, veja os Primeiros Passos.
Primeiros passos
Para ativar seu usuário no HPCC Marvin, é necessário fazer um primeiro acesso via ssh (Secury SHell), protocolo de rede seguro que permite a comunicação com servidores remotos.
Primeiro acesso 🚪
O primeiro acesso ao HPC Marvin é feito através do terminal ![]() (Linux ou MacOS) ou do PowerShell
(Linux ou MacOS) ou do PowerShell ![]() (Windows). Para isso, use o seguinte comando:
(Windows). Para isso, use o seguinte comando:
ssh <seu.login.cnpem>@marvin.cnpem.br
marie.curie@lnbio.cnpem.br. Logo, seu usuário é marie.curie. Sempre que encontrar <seu.login.cnpem>, digite marie.curie.
Quando solicitado, digite sua senha institucional.
Você pode receber um aviso solicitando sua confirmação antes de continuar conectando.
[...] Are you sure you want to continue connecting (yes/no/[fingerprint])?
Digite yes e pressione enter. Se tudo correu bem, você verá o cursor piscando no terminal, com um texto semelhante a:
[<seu.login.cnpem>@marvin ~]$
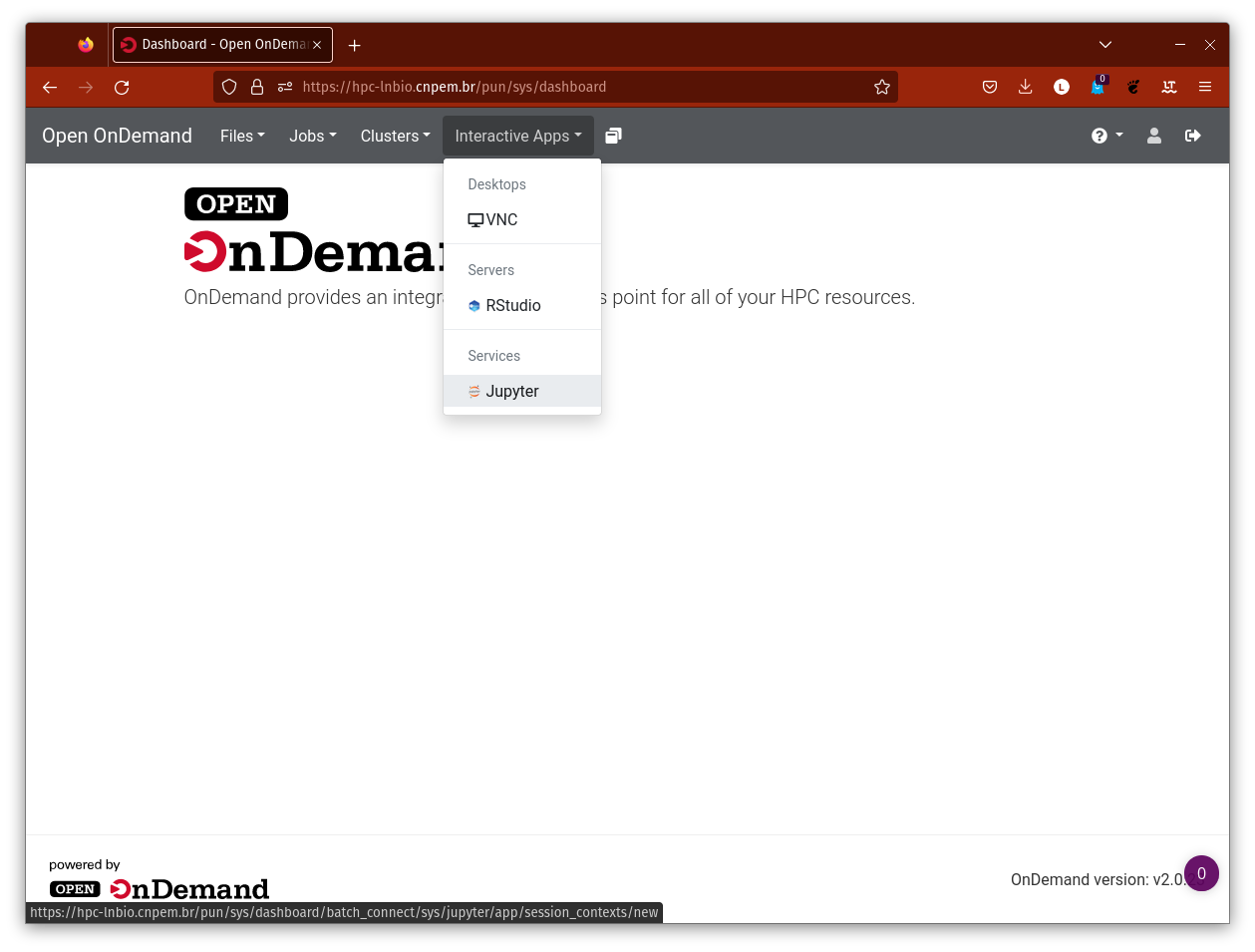
Após o primeiro login, você já poderá ler e gravar arquivos na aba Files do Open OnDemand (OOD), porém ainda não terá permissão para criar jobs, submeter tarefas ao SLURM ou utilizar os Interactive Apps.
Essa autorização é concedida manualmente. Para solicitá-la, registre um chamado na [LNBio] Suporte EDB do Jira em HPCC Marvin: Suporte ao usuário.
Se estiver no Windows e receber o seguinte erro, solicite ao TIC para instalar o ssh ou tente usar outro computador.
ssh: O termo 'ssh' não é reconhecido como nome de cmdlet, função, arquivo de script
ou programa operável. Verifique a grafia do nome ou, se um caminho tiver sido incluído,
veja se o caminho está correto e tente novamente.
Na linha:1 caractere:1
+ ssh marie.curie@marvin.cnpem.br
+ ~~~
+ CategoryInfo : ObjectNotFound (ssh:String) [], CommandNotFoundException
+ FullyQualifiedErrorId : CommandNotFoundException
Acesso pelo navegador 
Para acessar o HPCC Marvin pelo navegador, abra seu navegador e acesse:
https://marvin.cnpem.br
Lembre-se que este endereço só funcionará na rede interna do CNPEM. Para acessá-lo de fora do centro, é necessário usar a VPN. Caso não tenha este acesso à VPN, entre em contato com o DTI.
Na tela de login, use seu usuário (sem @lnbio.cnpem.br) e senha institucional.
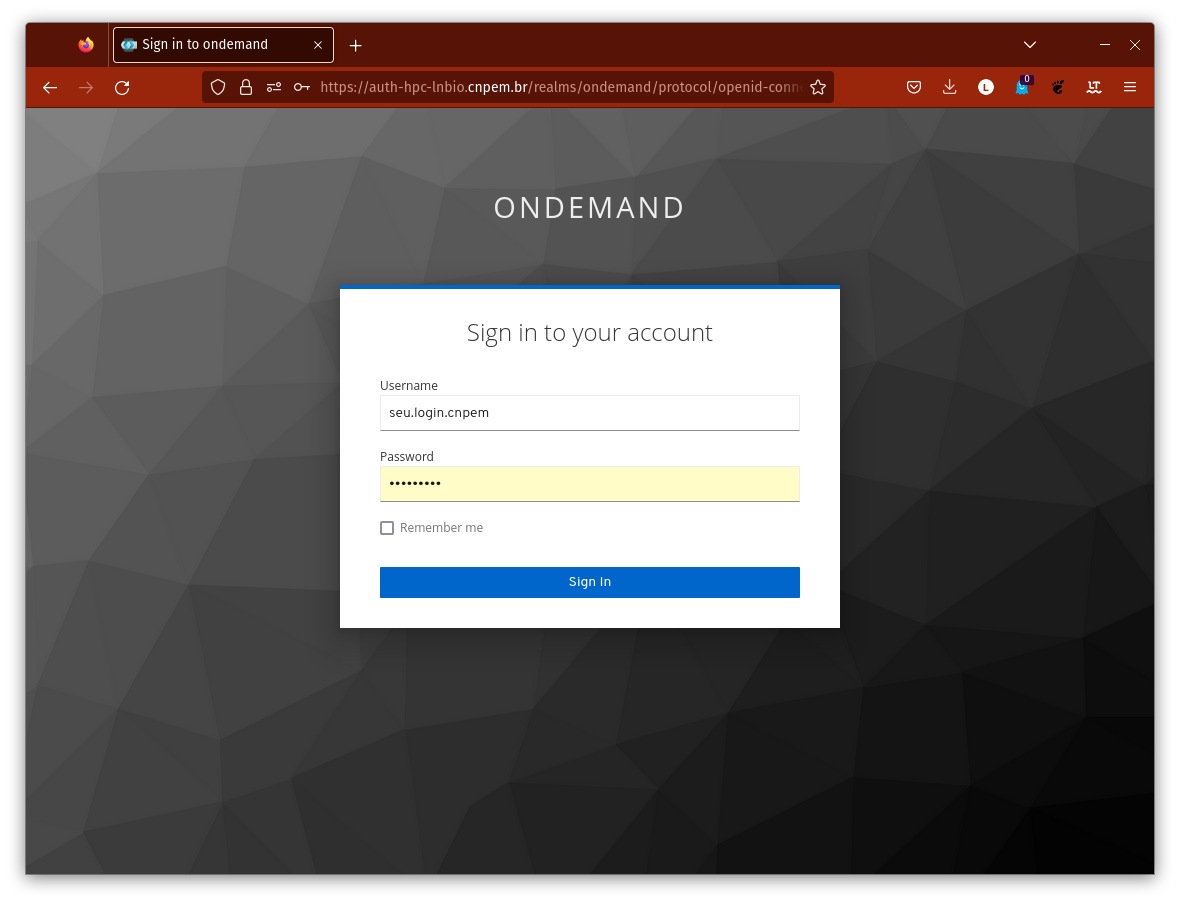
Após o login, você verá a interface principal do Open OnDemand:
Vídeo resumo
Abaixo, você pode ver um vídeo que resume os primeiros acesso ao HPCC Marvin, tanto pelo terminal quanto pelo navegador.
Se você tiver alguma dúvida ou precisar de ajuda, não hesite em entrar em contato com a equipe de suporte do sistema.
Arquitetura
Essa seção apresenta a arquitetura do HPCC Marvin, com informações detalhadas sobre:
Hardware
O hardware do HPCC Marvin foi adquirido em 2022 da empresa Atos, com investimento aproximado de US$350000. O cluster está fisicamente instalado no Data Center do Sirius/LNLS, contando com infraestrutura de refrigeração e energia.
O cluster é composto por um conjunto de servidores, chamados de nós, organizados conforme a topologia tradicional de um cluster HPC:
01nó de login (head node): ponto de entrada dos usuários, onde comandos são executados e jobs são submetidos.01nó de computação em CPU (CPU node): dedicado à execução de jobs que requerem alto desempenho de processamento em CPU.01nó de computação em GPU (GPU node): dedicado à execução de jobs que se beneficiam de aceleração por GPU.01sistema de armazenamento de alta performance (high performance storage system): sistema de arquivos compartilhado baseado em Lustre, voltado para I/O paralelo de alta velocidade.
As especificações técnicas de cada nó estão apresentadas na tabela a seguir:
| Nó | CPU | RAM | GPU | Armazenamento |
|---|---|---|---|---|
| Login | AMD EPYC 7352 24-Core @ 2.4 GHz | 256 GB | NVIDIA A40 (48 GB) | N/A |
| CPU | AMD EPYC 7742 64-Core @ 2.25 GHz | 1 TB | N/A | N/A |
| GPU | AMD EPYC 7742 64-Core @ 2.25 GHz | 2 TB | 8x NVIDIA A100 (40 GB) | N/A |
| Storage HPC | N/A | N/A | N/A | 300 TB |
Sistema Operacional
O HPCC Marvin utiliza o sistema operacional Rocky Linux 8.5, que é configurado de forma padronizada em todos os nós do HPCC, incluindo o nó de login, nós de computação (CPU e GPU) e o nó de Storage HPC.
Sistema de filas
Em ambientes HPC, é comum que múltiplos usuários estejam logados e executando jobs simultaneamente. Para gerenciar eficientemente a alocação de recursos (CPU, GPU, memória, etc) e a ordem de execução desses jobs, são usados sistemas de gerenciamento de filas.
O gerenciador de filas usado é o SLURM v21.08.8-2, que organiza a execução por meio de filas, chamadas de partitions, que armazenam os jobs submetidos pelos usuários. Assim que os recursos solicitados estão disponíveis, o SLURM inicia a execução dessas tarefas de forma automática.
Todos os jobs devem ser submetidos através do SLURM.
As filas de execução do HPCC Marvin são:
| Fila | Tempo limite | cpus-per-task (limite) | mem-per-cpu (default) | mem-per-cpu (limite) | GPU |
|---|---|---|---|---|---|
| debug-cpu | 30 minutos | 2 | 1GB | 2GB | Não |
| gui-cpu | 12 horas | 8 | 1GB | 4GB | Não |
| short-cpu | 5 dias | 64 | 1GB | 4GB | Não |
| long-cpu | 15 dias | 32 | 1GB | 4GB | Não |
| debug-gpu-small | 30 minutos | 2 | 1GB | 2GB | Sim (5GB) |
| gui-gpu-small | 12 horas | 8 | 1GB | 4GB | Sim (5GB) |
| short-gpu-small | 5 dias | 64 | 1GB | 8GB | Sim (5GB) |
| long-gpu-small | 15 dias | 32 | 1GB | 8GB | Sim (5GB) |
| debug-gpu-big | 30 minutos | 2 | 1GB | 2GB | Sim (40GB) |
| gui-gpu-big | 12 horas | 8 | 1GB | 4GB | Sim (40GB) |
| short-gpu-big | 5 dias | 64 | 1GB | 8GB | Sim (40GB) |
| long-gpu-big | 15 dias | 32 | 1GB | 8GB | Sim (40GB) |
Gerenciamento de jobs
Os jobs no HPCC Marvin são administrados pelo gerenciador de recursos computacionais SLURM (Simple Linux Utility for Resource Management) v21.08.8-2.
Essa seção apresenta infomações sobre gerenciamento de jobs, com informações detalhadas sobre:
- O que é um job?
- O que é SLURM?
- Verificando recursos
- Submissão de jobs
- Monitoramento de jobs
- Cancelamento de jobs
- Boas práticas
Referências adicionais
- Documentação: https://slurm.schedmd.com/documentation.html
- Tutorial: https://slurm.schedmd.com/tutorials.html
- Manual: https://slurm.schedmd.com/man_index.html
- FAQ: https://slurm.schedmd.com/faq.html
O que é SLURM?
O SLURM (Simple Linux Utility for Resource Management) é um sistema de gerenciamento de filas de código aberto projetado para organizar o acesso aos recursos computacionais em um cluster. Ele permite que múltiplos usuários compartilhem os recursos do cluster de forma eficiente.
O que é um job?
Um job é uma tarefa computacional submetida para execução no cluster, como scripts, programas, simulações ou qualquer outro tipo de processamento.
No HPCC Marvin, os jobs são enviados ao SLURM por meio de scripts de submissão. O SLURM insere os jobs em uma fila e os executa conforme a disponibilidade dos recursos, seguindo políticas de agendamento definidas.
Cada job pode variar em complexidade: desde a execução de um único comando até fluxos compostos por múltiplas etapas e dependências. Os usuários podem especificar requisitos como número de CPUs, quantidade de memória, uso de GPUs e tempo estimado de execução.
Os jobs podem ser executados em segundo plano (sbatch) ou de forma interativa (srun --pty bash -i), conforme a necessidade. O SLURM também oferece recursos avançados, como monitoramento em tempo real, controle de dependências e suporte à retomada em caso de falhas.
Submissão de jobs
Para submeter um job, você deve criar um script de submissão com os parâmetros adequados e usar o comando sbatch:
sbatch teste.sh
O script de submissão define as características do trabalho, como nome, partição, número de nós e CPUs, memória, tempo máximo de execução, e arquivos de saída. Um exemplo básico:
#!/bin/bash
#SBATCH --job-name=teste
#SBATCH --partition=debug-cpu
#SBATCH --cpus-per-task=1
#SBATCH --mem=1G
#SBATCH --time=00:10:00
#SBATCH --output=output_%j.log
echo "Olá do cluster Marvin!"
hostname
date
sleep 60
echo "Trabalho concluído."
Indicando a partição do SLURM (fila)
Para especificar uma partição (fila) use:
#SBATCH --partition=<substitua pelo nome da partição>
Cada partição ou fila possui recursos e limites diferentes, elas podem ser consultadas em Sistema de filas.
Solicitando recursos específicos
- Solicite o número de CPUs para a tarefa
#SBATCH --cpus-per-task=<substitua pelo numero de cpus>
- A solicitação de GPUs varia de acordo com a partição. Partições
*-gpu-smallpossuem GPUs de 5GB de memória, enquanto que partições*-gpu-bigpossuem GPUs A100 com 40GB de memória.
Para as filas *-gpu-small use:
#SBATCH --gres=gpu:1g.5gb:1
Para as filas *-gpu-big use:
#SBATCH --gres=gpu:a100:1
- A solicitação da quantidade de memória pode ser feita em quantidade total para o job ou quantidade por cpu.
Para solicitar a quantidade total use:
#SBATCH --mem=16G
Para solicitar a quantidade por CPU use:
#SBATCH --mem-per-cpu=4GB
- Solicitação de tempo
Para um melhor funcionamento do sistema gerenciador de tarefas e recursos (SLURM) é importante que o usuário indique o tempo aproximado de execução da tarefa, mesmo sendo sobreestimado. Através desse tempo, o SLURM conseguirá alocar jobs em janelas ociosas e otimizar o uso dos recursos.
Exemplo solicitando 10 horas de tempo de processamento.
#SBATCH --time=10:00:00
Trabalhos interativos
Você pode iniciar uma sessão interativa com:
srun -p debug-cpu --pty bash
Ou com recursos definidos:
srun -p short-gpu-small --gres gpu:1g.5gb:1 -c 4 --mem 8G --time 01:00:00 --pty bash
Monitoramento de jobs
Para acompanhar o status dos seus jobs, você pode usar o comando squeue:
squeue -u $USER
Para obter informações detalhadas de um job específico, use o comando scontrol:
scontrol show job <job_id>
Substitua
<job_id> pelo ID do seu job.Cancelamento de jobs
Para cancelar um job específico, use o comando scancel seguido do ID do job:
scancel <jon_id>
Substitua
<job_id> pelo ID do seu job.Para cancelar todos os seus jobs em execução ou na fila, use o seguinte comando:
scancel -u $USER
Sempre verifique o status do seus jobs antes de cancelá-los.
Verificando recursos disponíveis
O SLURM fornece ferramentas para verificar a disponibilidade de recursos no cluster. Para visualizar as partições disponíveis e seus limites, você pode usar o comando:
sinfo
Esse comando exibe informações sobre as partições, incluindo o número de nós disponíveis, o número de nós ocupados e o estado atual de cada partição.
Para visualizar informações detalhadas sobre os nós, incluindo o estado de cada nó, você pode usar:
sinfo -N -l
Esse comando fornece uma visão detalhada de cada nó, incluindo informações sobre a memória, CPUs e o estado atual.
Boas práticas
Para garantir o uso eficiente do sistema e evitar problemas durante a execução de jobs, siga estas boas práticas:
-
Especifique os recursos necessários: Solicite apenas o que for necessário para evitar desperdício e facilitar o agendamento.
-
Defina um tempo limite adequado: Um tempo muito curto pode interromper seu job; muito longo pode atrasar a fila.
-
Utilize o arquivo de saída: Monitore a execução do job e facilite a depuração de erros.
-
Faça testes com jobs menores: Teste seu pipeline com dados ou tempos reduzidos antes de escalar para execuções maiores.
-
Evite sobrecarga de I/O: Reduza o número de acessos simultâneos ao sistema de arquivos compartilhado sempre que possível.
-
Não processar dados no nó de login: Execute seus scripts e comandos de processamento de dados apenas dentro de um job para evitar sobrecarga no nó de login.
Gerenciamento de dados
Os dados são gerenciados a nível de usuário no HPCC Marvin.
Essa seção apresenta informações sobre gerenciamento de dados, com informações detalhadas sobre:
Armazenamento de dados
O HPCC Marvin fornece duas opções principais de armazenamento de dados: pasta pessoal e pasta compartilhada por grupos de pesquisa.
Pasta pessoal
Cada usuário do HPCC Marvin tem acesso a um espaço de armazenamento pessoal, onde pode armazenar e compartilhar seus dados.
marie.curie, sua pasta estará localizado em /home/marie.curie. Você acessa sua pasta pessoal ao fazer login no sistema, e ela é automaticamente montada como seu diretório inicial ($HOME).
Esta pasta (e.g., seu HOME) é exclusiva para cada usuário e é utilizado para guardar arquivos, scripts, resultados e dados intermediários necessários apenas para suas atividades de pesquisa.
Pasta compartilhada por grupos de pesquisa
Os grupos de pesquisa podem solicitar uma pasta compartilhada para armazenar dados que precisam ser acessados por vários membros do grupo.
/shared/groups/<sigla-do-grupo>.
Para solicitar uma pasta compartilhada, registre um chamado na [LNBio] Suporte EDB do Jira em HPCC Marvin: Suporte ao usuário. Informe o nome do grupo de pesquisa e a sigla que deseja (/shared/groups/<sigla-do-grupo>).
Transferência de dados
O HPCC Marvin oferece várias opções para transferir dados entre o sistema e seu computador local. As principais ferramentas para essa tarefa são: SFTP, SCP e Rsync.
SSH File Transfer Protocol (SFTP)
O SFTP é um protocolo seguro que permite a transferência de arquivos entre sistemas operacionais diferentes. É útil quando você precisa transferir arquivos de forma segura. Para usar o SFTP, execute o seguinte comando:
sftp <seu.login.cnpem>@marvin.cnpem.br
Após se conectar, você pode usar comandos como:
ls: lista os arquivos no diretório atual;cd: navega para um diretório específico;put: envia um arquivo do seu computador local para o HPCC Marvin;get: baixa um arquivo do HPCC Marvin para o seu computador local.
Secure Copy Protocol (SCP)
O SCP (Secure Copy Protocol) é outro protocolo seguro que usa a criptografia SSH para transferir arquivos. Ele é semelhante ao SFTP, mas é mais simples de usar e não tem recursos de navegação.
Para transferir um arquivo (e.g., file.txt) do seu computador local para o HPCC Marvin, use o comando:
scp file.txt <seu.login.cnpem>@marvin.cnpem.br:/caminho/de/destino/
Para transferir um diretório (e.g., directory/) do seu computador local para o HPCC Marvin, use o comando:
scp -r directory/ <seu.login.cnpem>@marvin.cnpem.br:/caminho/de/destino/
Para transferir um arquivo (e.g., file.txt) do HPCC Marvin para o seu computador local, use o comando:
scp <seu.login.cnpem>@marvin.cnpem.br:/caminho/do/arquivo/file.txt /caminho/local/de/destino/
Para transferir um diretório (e.g., directory/) do HPCC Marvin para o seu computador local, use o comando:
scp -r <seu.login.cnpem>@marvin.cnpem.br:/caminho/do/diretorio/directory/ /caminho/local/de/destino/
Rsync
O Rsync é um protocolo de transferência de arquivos que pode sincronizar diretórios entre hosts. Ele usa uma conexão segura SSH e é útil para transferir grandes quantidades de dados ou sincronizar arquivos entre sistemas.
Para transferir um arquivo (e.g., file.txt) do seu computador local para o HPCC Marvin, use o comando:
rsync -avz file.txt <seu.login.cnpem>@marvin.cnpem.br:/caminho/de/destino/
Para transferir um diretório (e.g., directory/) do seu computador local para o HPCC Marvin, use o comando:
rsync -avz directory <seu.login.cnpem>@marvin.cnpem.br:/caminho/de/destino/
Para transferir um arquivo (e.g., file.txt) do HPCC Marvin para o seu computador local, use o comando:
rsync -avz <seu.login.cnpem>@marvin.cnpem.br:/caminho/do/arquivo/file.txt /caminho/local/de/destino/
Para transferir um diretório (e.g., directory/) do HPCC Marvin para o seu computador local, use o comando:
rsync -avz <seu.login.cnpem>@marvin.cnpem.br:/caminho/do/diretorio/directory/ /caminho/local/de/destino/
-a mantém as permissões de arquivos, a flag -v mostra o progresso da transferência e a flag -z comprime os dados antes de transferi-los.
Compartilhamento de dados
Os dados são compartilhados a nível de usuário no HPCC Marvin, usando o sistema de controle de acesso do Linux, conhecido como Access Control Lists (ACLs). As ACLs permitem que você defina permissões específicas para usuários e grupos em diretórios e arquivos, facilitando o compartilhamento seguro de dados entre usuários.
Para garantir a segurança e a privacidade dos dados, cada diretório de usuário no HPCC Marvin possui permissão 700 ou u:rwx, g:---, o:---. Isso significa que apenas o próprio usuário “u” tem permissão de leitura “r”, escrita w e execução x, enquanto membros do grupo g e outros usuários o não têm acesso.
No entanto, em alguns casos, você pode precisar permitir o acesso a determinados diretórios, como para compartilhar um projeto com outros usuários.
Usando o ACL para compartilhar dados
Nesta seção, vamos usar o usuário hipotético marie.curie como exemplo para demonstrar como compartilhar dados com outros usuários no HPCC Marvin.
Verificando as permissões
Para verificar as permissões de um arquivo ou um diretório específico, use o seguinte comando no terminal:
getafcl /caminho/do/diretorio_ou_arquivo
Por exemplo, para verificar as permissões do seu HOME, você pode usar:
$ getafcl /home/marie.curie
# file: /home/marie.curie
# owner: marie.curie
# group: domain\040users
user::rwx
group::---
other::---
Concedendo permissão de acesso a outros usuários
Primeiramente, para compartilhar um arquivo ou diretório com outros usuários, você precisa começar alterarando as permissões do diretório $HOME para 711. Para isso, use o comando:
chmod 711 /home/marie.curie
Então, cheque as mudanças com o comando getfacl:
$ getfacl /home/marie.curie
# file: /home/marie.curie
# owner: marie.curie
# group: domain\040users
user::rwx
group::--x
other::--x
Em seguida, você pode usar o comando setfacl para adicionar permissões de acesso a outros usuários. Por exemplo, para conceder permissão de leitura ao usuário “joao.guerra” no diretório “pasta_compartilhada”, use o comando:
setfacl -m u:joao.guerra:r-x /home/marie.curie/pasta_compartilhada
x foi adicionado para que o usuário consiga executar o comando ls e listar os arquivos dentro do diretório compartilhado.
Então, cheque as mudanças com o comando getfacl:
$ getfacl /home/marie.curie/pasta_compartilhada
# file: /home/marie.curie/pasta_compartilhada
# owner: marie.curie
# group: domain\040users
user::rwx
user:joao.guerra:r-x
group::--x
mask::r-x
other::--x
Caso você queira dar permissões de acesso recursivamente para todos os arquivos e subdiretórios dentro de “pasta_compartilhada”, você pode usar a opção -R:
setfacl -R -m u:joao.guerra:r-x /home/marie.curie/pasta_compartilhada
Caso você queira conceder acesso para todas os arquivos e subdiretórios que podem ser criados dentro de “pasta_compartilhada”, você pode usar a opção -d:
setfacl -d -m u:joao.guerra:r-x /home/marie.curie/pasta_compartilhada
Então, cheque novamente as mudanças com o comando getfacl:
$ getfacl /home/marie.curie/pasta_compartilhada
# file: /home/marie.curie/pasta_compartilhada
# owner: marie.curie
# group: domain\040users
user::rwx
user:joao.guerra:r-x
group::--x
mask::r-x
other::--x
default:user::rwx
default:user:joao.guerra:r-x
default:group::--x
default:mask::r-x
default:other::--x
Para adicionar mais permissões, você pode usar outras opções como w para escrita e x para execução. Por exemplo, para dar ao usuário “joao.guerra” permissão de escrita e execução na pasta “pasta_compartilhada”, use o comando:
setfacl -m u:joao.guerra:rwx /home/marie.curie/pasta_compartilhada
Então, cheque novamente as mudanças com o comando getfacl:
$ getfacl /home/marie.curie/pasta_compartilhada
# file: /home/marie.curie/pasta_compartilhada
# owner: marie.curie
# group: domain\040users
user::rwx
user:joao.guerra:rwx
group::--x
mask::rwx
other::--x
default:user::rwx
default:user:joao.guerra:r-x
default:group::--x
default:mask::r-x
default:other::--x
Removendo permissões de acesso
Para remover as permissões de acesso de um usuário específico, utilize o comando setfacl -x seguido do usuário desejado. Por exemplo, para remover as permissões de acesso do usuário “joao.guerra” no diretório “pasta_compartilhada”, use o comando:
setfacl -x u:joao.guerra /home/marie.curie/pasta_compartilhada
Então, cheque novamente as mudanças com o comando getfacl:
$ getfacl /home/marie.curie/pasta_compartilhada
# file: /home/marie.curie/pasta_compartilhada
# owner: marie.curie
# group: domain\040users
user::rwx
group::--x
other::--x
default:user::rwx
default:user:joao.guerra:r-x
default:group::--x
default:mask::r-x
default:other::--x
Caso você queira remover permissões de acesso recursivamente para todos os arquivos e subdiretórios dentro de “pasta_compartilhada”, você pode usar a opção -R:
setfacl -R -x u:joao.guerra /home/marie.curie/pasta_compartilhada
Caso você queira remover acesso para todas os arquivos e subdiretórios que podem ser criados dentro de “pasta_compartilhada”, você pode usar a opção -d:
setfacl -d -x u:joao.guerra /home/marie.curie/pasta_compartilhada
Então, cheque novamente as mudanças com o comando getfacl:
$ getfacl /home/marie.curie/pasta_compartilhada
# file: /home/marie.curie/pasta_compartilhada
# owner: marie.curie
# group: domain\040users
user::rwx
group::--x
mask::--x
other::--x
default:user::rwx
default:group::--x
default:mask::--x
default:other::--x
Para remover todas as permissões de acesso, você pode usar o comando setfacl -b seguido do caminho do diretório. Por exemplo, para remover todas as permissões de acesso no diretório “pasta_compartilhada”, use o comando:
setfacl -b /home/marie.curie/pasta_compartilhada
Então, cheque novamente as mudanças com o comando getfacl:
$ getfacl /home/marie.curie/pasta_compartilhada
# file: /home/marie.curie/pasta_compartilhada
# owner: marie.curie
# group: domain\040users
user::rwx
group::--x
other::--x
Usando links simbólicos para pastas compartilhadas
Para facilitar o acesso a pastas compartilhadas, você pode criar links simbólicos (atalhos) para essas pastas em seu HOME. Isso é especialmente útil quando você precisa acessar frequentemente uma pasta compartilhada sem precisar navegar até o caminho completo toda vez.
Para criar um link simbólico, você pode usar o comando ln -s seguido do caminho da pasta compartilhada e do caminho onde deseja criar o link. Por exemplo, se você quiser criar um link simbólico para a pasta compartilhada /shared/groups/edb no seu diretório HOME, você pode usar o seguinte comando:
ln -s /shared/groups/edb /home/marie.curie/edb
singularity run --bind /caminho/da/pasta/compartilhada:/caminho/dentro/do/container imagem.sif.
Referências adicionais
- An introduction to Linux Access Control Lists (ACLs)
- Red Hat Docs - Chapter25. Managing file permissions
- Red Hat Docs - Chapter 28. Managing the Access Control List
- Arch Linux Wiki - Access Control Lists (Português)
- Using ACLs
Princípios FAIR
Os princípios FAIR são diretrizes para tornar os dados de pesquisa mais acessíveis, interoperáveis e reutilizáveis. FAIR é um acrônimo que significa:
- Findable: Os dados devem ser fáceis de encontrar por humanos e máquinas.
- Accessible: Os dados devem ser acessíveis de forma clara e transparente, com informações sobre como obtê-los.
- Interoperable: Os dados devem ser compatíveis com outros conjuntos de dados e sistemas, permitindo a integração e o uso conjunto.
- Reusable: Os dados devem ser descritos de forma que possam ser reutilizados em diferentes contextos e por diferentes usuários.
Gerenciamento de ambiente
O HPCC Marvin oferece flexibilidade para que os usuários configurem seus próprios ambientes de desenvolvimento. O gerenciamento de ambientes é feito por meio do sistema de módulos (Lmod), o que permite carregar, combinar e personalizar bibliotecas conforme as necessidades de cada projeto.
Para visualizar os módulos disponíveis, utilize:
module avail
Para listar os módulos com a descrição, utilize:
module spider
Para carregar um módulo específico, use:
module load <nome>/<versão>
Quando múltiplas versões de um software estão disponíveis, uma delas é definida como padrão (indicada por
(D)).Para listar os módulos atualmente carregados:
module list
Ambiente de desenvolvimento
Além dos módulos pré-instalados, os usuários podem criar e gerenciar seus próprios ambientes com ferramentas como:
- miniforge: Ambiente e gerenciamento de pacotes para Python, R, C/C++ e outras linguagens. Para habilitar, carregue o módulo
miniforge:
module load miniforge
- uv: Gerenciador de ambientes Python extremamente rápido e leve, compatível com
pipepyproject.toml. Para habilitar, carregue o módulouv:
module load uv
Compiladores
O Marvin oferece suporte para desenvolvimento de aplicações em C, C++, Fortran e outros, com os seguintes compiladores e ferramentas de build disponíveis no ambiente padrão:
| Ferramenta | Descrição |
|---|---|
gcc | Compilador GNU para C |
g++ | Compilador GNU para C++ |
gfortran | Compilador GNU para Fortran |
clang | Compilador LLVM para C |
clang++ | Compilador LLVM para C++ |
make | Gerenciador de build tradicional |
cmake | Sistema de build multiplataforma |
Boas práticas
Para garantir um ambiente de desenvolvimento eficiente e organizado, recomenda-se:
- Criar ambientes virtuais reutilizáveis com
uveminiforge; - Manter os ambientes organizados, evitando a criação de múltiplos ambientes redundantes;
- Documentar dependências em arquivos como
README.md,pyproject.toml(uv/pip),requirements.txt(uv/pip) e/ouenvironment.yml(conda/mamba), facilitando o compartilhamento e a reprodução do ambiente por outros usuários.
Aplicativos e Programas
No HPCC Marvin, os aplicativos e programas são disponibilizados principalmente por meio do sistema de módulos (Lmod), permitindo que os usuários carreguem e utilizem diferentes versões conforme suas necessidades. Eventualmente, alguns podem estar disponíveis fora deste padrão, quando há necessidades específicas ou limitações técnicas.
Para listar os módulos disponíveis, utilize o comando:
module avail
Para listar os módulos com a descrição, utilize:
module spider
Para carregar um módulo específico, use:
module load <nome>/<versão>
Quando múltiplas versões de um software estão disponíveis, uma delas é definida como padrão (indicada por
(D)).Para listar os módulos carregados na sua sessão, utilize:
module list
Para solicitar a instalação ou atualização de um aplicativo ou programa, registre um chamado [LNBio] Suporte EDB do Jira em HPCC Marvin: Aplicativos, Programas e Sistemas.
Bioimagens
Os programas e aplicativos relacionados à processamento e análise de imagens biológicas são:
Biologia Estrutural
Os programas e aplicativos relacionados à modelagem, dinâmica molecular, predição e análise estrutural de proteínas são:
Descoberta de fármacos (Drug Discovery)
Os programas e aplicativos relacionados à descoberta e desenvolvimento de fármacos são:
HiC
Os programas e aplicativos relacionados à análise, processamento e visualização de dados de interação cromossômica (Hi-C) são:
Microscopia Eletrônica
Os programas e aplicativos relacionados a processamento, análise e visualização de microscopia eletrônica são:
Ômicas
Os programas e aplicativos relacionados a processamento, análise e visualização de ômicas são:
3dmod
O 3dmod é um programa de visualização e modelagem de dados de microscopia eletrônica em 3D. Ele faz parte do pacote IMOD, que é amplamente utilizado para a análise e reconstrução de imagens de tomografia eletrônica.
Para mais informações sobre o 3dmod, acesse https://bio3d.colorado.edu/imod/doc/3dmodguide.html.
Carregando o módulo
O 3dmod está disponível dentro do módulo scipion/3.8.3. Para utilizá-lo no HPCC Marvin, você deve carregar o módulo scipion/3.8.3:
module load scipion/3.8.3
Para acessar a documentação do modulo, utilize:
module help scipion/3.8.3
Como executar o 3dmod no Open OnDemand
Para executar o 3dmod, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o 3dmod:
# Habilitar o módulo
module load scipion/3.8.3
# Iniciar o 3dmod com interface gráfica
3dmod
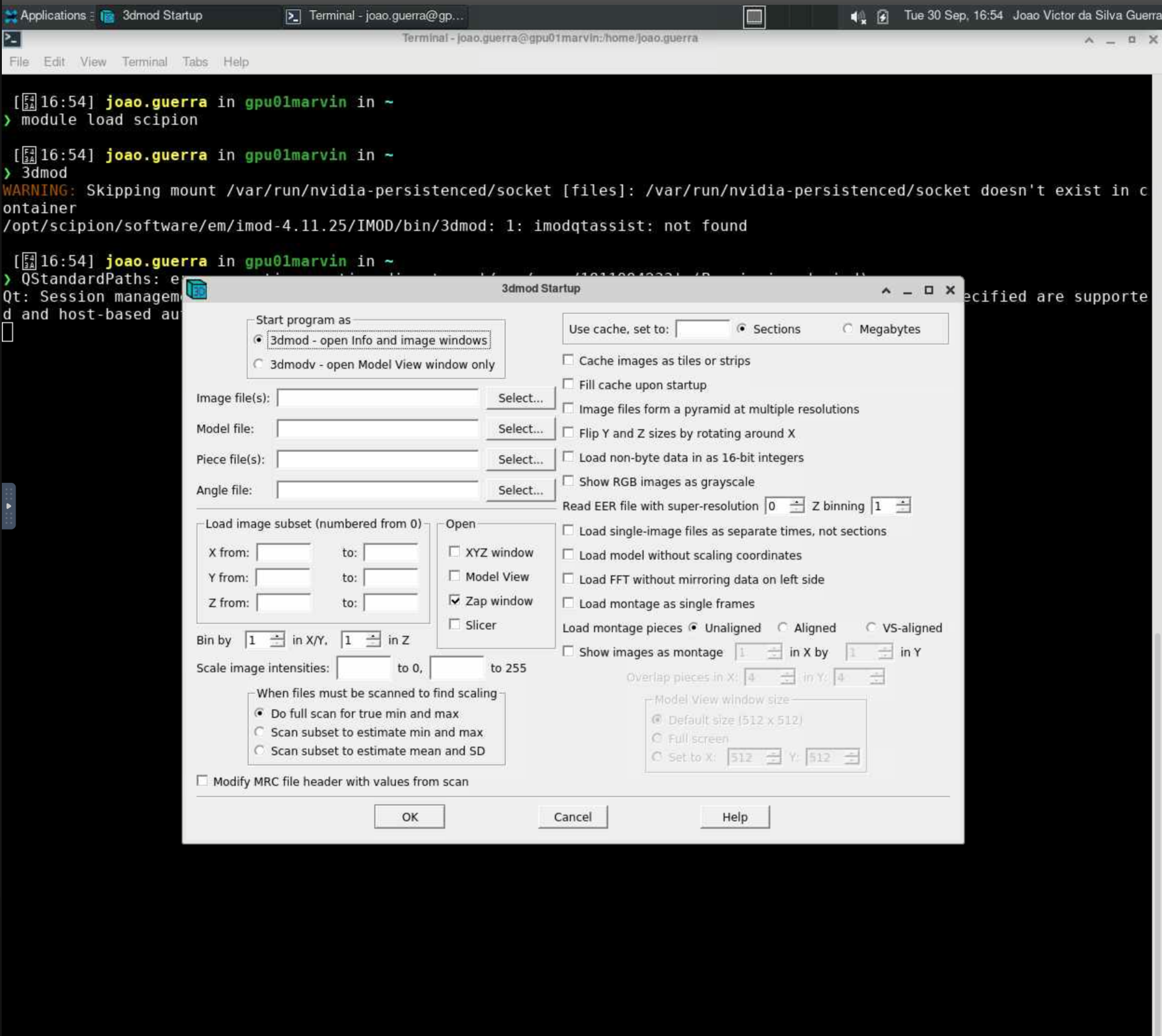
Para mais detalhes sobre os parâmetros do 3dmod, use:
3dmod -h
3D Slicer
O 3D Slicer é uma plataforma de código aberto para análise e visualização de imagens médicas em 3D. Ele oferece ferramentas avançadas para segmentação, reconstrução, registro e modelagem anatômica, sendo amplamente utilizado em pesquisa biomédica, planejamento cirúrgico e aplicações clínicas.
Para mais informações, consulte a documentação oficial do 3D Slicer: https://slicer.readthedocs.io/en/latest/.
Carregando o módulo
Para habilitar o 3D Slicer no Marvin, você deve carregar o módulo 3dslicer
module load 3dslicer/5.8.1
Para acessar a documentação do modulo, utilize:
module help 3dslicer/5.8.1
Como executar o 3D Slicer no Open OnDemand
Para executar o 3D Slicer, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o 3D Slicer:
# Habilitar o módulo
module load 3dslicer/3.8.3
# Iniciar o 3dslicer com interface gráfica
Slicer
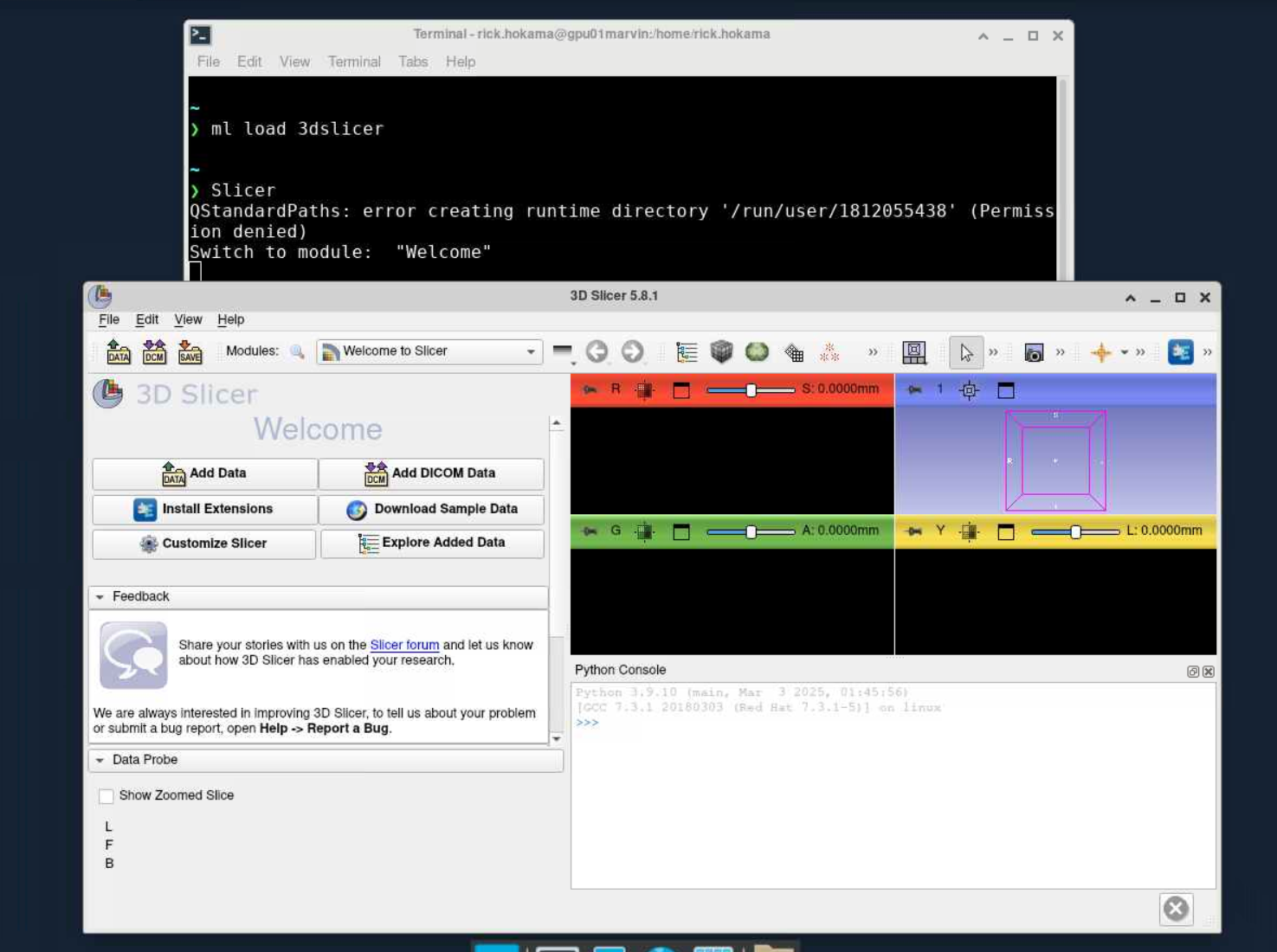
Para mais detalhes sobre os parâmetros do 3D Slicer, use:
Slicer --help
AlphaFold
O AlphaFold é um programa de modelagem de estruturas proteicas utilizando redes neurais artificiais (Deep Learning). Além de proteínas individuais, ele também permite modelar multímeros e complexos.
Para mais informações sobre o AlphaFold, acesse https://github.com/deepmind/alphafold/.
Carregando o módulo
Para habilitar o AlphaFold no HPCC Marvin, você deve carregar o módulo alphafold:
module load alphafold
As versões disponíveis do AlphaFold no HPCC Marvin são:
alphafold/2.3.2 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help alphafold
Submetendo jobs
A execução do AlphaFold no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo alphafold.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=alphafold2
#SBATCH --partition=short-gpu-big
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=8
#SBATCH --gres=gpu:a100:1
#SBATCH --mem=64G
#SBATCH --time=24:00:00
module load alphafold/2.3.2
OUTPUT_DIR="resultado_af2"
FASTA_FILE="meu_target.fasta"
alphafold \\
--output_dir=$OUTPUT_DIR \\
--fasta_paths=$FASTA_FILE \\
--max_template_date=2023-11-01 \\
--model_preset=monomer_ptm \\
--db_preset=full_dbs
O
FASTA_FILE deve apontar para o arquivo FASTA da proteína que você deseja modelar. O OUTPUT_DIR é onde os resultados serão salvos.Para submeter o job, salve o script e utilize o comando sbatch:
sbatch alphafold.sh
Para mais detalhes sobre os parâmetros do AlphaFold, use:
alphafold --help
Boltz-2
O Boltz-2 é um modelo para predição de interação biomolecular. Boltz-2 vai além AlphaFold3 e Boltz-1 ao juntar modelagem de complexa estruturas e afinidade de ligantes, um componente crítico para o design molecular preciso.
Para mais informações sobre o Boltz-2, acesse https://github.com/jwohlwend/boltz/tree/main/docs.
Carregando o módulo
Para habilitar o Boltz-2 no HPCC Marvin, você deve carregar o módulo boltz:
module load boltz
As versões disponíveis do Boltz no HPCC Marvin são:
boltz/2.1.1boltz/2.2.0boltz/2.2.1 (D)
(D) indica a versão padrão.Para acessar a documentação do módulo, utilize:
module help boltz
Submetendo jobs
A execução do Boltz no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo boltz.sh, com o seguinte conteúdo:
#! /bin/bash
#SBATCH --job-name=boltz
#SBATCH --partition=short-gpu-big
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=8
#SBATCH --gres=gpu:a100:1
#SBATCH --mem=64G
#SBATCH --time=24:00:00
module load boltz
YAML_DIR="./yaml"
OUTPUT_DIR="./"
boltz \\
predict \\
$YAML_DIR \\
--use_potentials \\
--out_dir $OUTPUT_DIR
O
YAML_DIR deve apontar para o diretório que contém os arquivos.yaml ou ao .yaml da proteína que você deseja modelar ou predizer afinidade. O OUTPUT_DIR é onde os resultados serão salvos.Para submeter o job, salve o script e utilize o comando sbatch
sbatch boltz.sh
Para mais detalhes sobre parâmetros de configuração do boltz, use:
boltz predict --help
Para mais informações sobre configuração do yaml, cheque: https://github.com/jwohlwend/boltz/blob/main/docs/prediction.md
Exemplo de YAML de configuração:
sequences:
- protein:
id: A
sequence: ANPCCSNPCQNRGECMSTGFDQYKCDCTRTGFYGENCTTPEFLTRIKLLLKPTPNTVHYILTHFKGVWNIVNNIPFLRSLIMKYVLTSRSYLIDSPPTYNVHYGYKSWEAFSNLSYYTRALPPVADDCPTPMGVKGNKELPDSKEVLEKVLLRREFIPDPQGSNMMFAFFAQHFTHQFFKTDHKRGPGFTRGLGHGVDLNHIYGETLDRQHKLRLFKDGKLKYQVIGGEVYPPTVKDTQVEMIYPPHIPENLQFAVGQEVFGLVPGLMMYATIWLREHNRVCDILKQEHPEWGDEQLFQTSRLILIGETIKIVIEDYVQHLSGYHFKLKFDPELLFNQQFQYQNRIASEFNTLYHWHPLLPDTFNIEDQEYSFKQFLYNNSILLEHGLTQFVESFTRQIAGRVAGGRNVPIAVQAVAKASIDQSREMKYQSLNEYRKRFSLKPYTSFEELTGEKEMAAELKALYSDIDVMELYPALLVEKPRPDAIFGETMVELGAPFSLKGLMGNPICSPQYWKPSTFGGEVGFKIINTASIQSLICNNVKGCPFTSFNVQ
msa: ./msa/msa.a3m
- ligand:
id: B
smiles: COc1ccc(C2C(=O)c3ccccc3C2=O)cc1
constraints:
- pocket:
binder: B
contacts:
- - A
- 492
- - A
- 318
max_distance: 6
force: true
properties:
- affinity:
binder: B
Cellranger
O Cellranger é um conjunto de pipelines de análise desenvolvido pela 10x Genomics para processar dados de sequenciamento de célula única (single-cell). Ele realiza o alinhamento de leituras, atribuição de barcodes, contagem de identificadores moleculares únicos (UMIs) e análise de expressão gênica.
Para mais informações e documentação completa, acesse: https://www.10xgenomics.com/support/software/cell-ranger/latest/getting-started.
Carregando o módulo
Para habilitar o Cellranger no HPCC Marvin, você deve carregar o módulo cellranger:
module load cellranger
As versões disponíveis do cellranger no HPCC Marvin são:
cellranger/10.0.0 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help cellranger
Executando o módulo
Modelo de uso do Cellranger via linha de comando:
cellranger <COMMAND>
Ao executar cellranger -h, é possível consultar a lista de comandos disponíveis:
cellranger 10.0.0
Process 10x Genomics Gene Expression, Feature Barcode, and Immune Profiling data
Usage: cellranger <COMMAND>
Commands:
count Count gene expression and/or feature barcode reads from a single sample and GEM well
multi Analyze multiplexed data or combined gene expression/immune profiling/feature barcode data
multi-template Output a multi config CSV template
vdj Assembles single-cell VDJ receptor sequences from 10x Immune Profiling libraries
aggr Aggregate data from multiple Cell Ranger runs
annotate Annotate cell-types from outputs of a CellRanger run
reanalyze Re-run secondary analysis (dimensionality reduction, clustering, etc)
mkvdjref Prepare a reference for use with CellRanger VDJ
mkfastq Run Illumina demultiplexer on sample sheets that contain 10x-specific sample index sets
testrun Execute the 'count' pipeline on a small test dataset
cloud Invoke cloud commands
mat2csv Convert a feature-barcode matrix to CSV format
mkref Prepare a reference for use with 10x analysis software. Requires a GTF and FASTA
mkgtf Filter a GTF file by attribute prior to creating a 10x reference
upload Upload analysis logs to 10x Genomics support
sitecheck Collect Linux system configuration information
telemetry Configure and inspect telemetry settings and data
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
Para mais detalhes sobre os parâmetros de cada comando do Cellranger, use:
cellranger help [COMMAND]
Arquivos de referência
O Cellranger requer arquivos de referência (como genomas, transcriptomas e índices de amostras) para a execução das análises. No HPCC Marvin, essas referências oficiais (obtidas diretamente da página de downloads do Cellranger) já estão centralizadas e prontas para uso no seguinte diretório: /public/cellranger.
Para otimizar o espaço do cluster e evitar redundâncias, não recomendamos o armazenamento de cópias de arquivos de referência em diretórios pessoais (como o seu /home). Caso a sua análise exija uma referência ou genoma customizado que ainda não esteja disponível no diretório público, por favor, entre em contato com a equipe da EDB.
Atualmente, essa é a estrutura dos dados de referência do Cellranger:
/public/cellranger
├── probe-sets
│ ├── barcode-sequence
│ │ └── flex-v2-384.txt
│ └── human-transcriptome
│ ├── Chromium_Human_Transcriptome_Probe_Set_v2.0.0_GRCh38-2024-A.bed
│ ├── Chromium_Human_Transcriptome_Probe_Set_v2.0.0_GRCh38-2024-A.csv
│ ├── Chromium_Human_Transcriptome_Probe_Set_v2.0.0_GRCh38-2024-A.offtarget.csv
│ └── Chromium_Human_Transcriptome_Probe_Set_v2.0.0_GRCh38-2024-A.probe_metadata.tsv
├── references
│ └── human
│ └── refdata-gex-GRCh38-2024-A
│ ├── fasta
│ │ ├── genome.fa
│ │ └── genome.fa.fai
│ ├── genes
│ │ └── genes.gtf.gz
│ ├── reference.json
│ └── star
│ ├── chrLength.txt
│ ├── chrNameLength.txt
│ ├── chrName.txt
│ ├── chrStart.txt
│ ├── exonGeTrInfo.tab
│ ├── exonInfo.tab
│ ├── geneInfo.tab
│ ├── Genome
│ ├── genomeParameters.txt
│ ├── SA
│ ├── SAindex
│ ├── sjdbInfo.txt
│ ├── sjdbList.fromGTF.out.tab
│ ├── sjdbList.out.tab
│ └── transcriptInfo.tab
└── sample-index-sequences
├── Chromium-i7-Multiplex-Kit-N-Set-A-sample-indexes-plate.csv
├── Chromium-i7-Multiplex-Kit-N-Set-A-sample-indexes-plate.json
├── chromium-shared-sample-indexes-plate-5.csv
├── chromium-shared-sample-indexes-plate-5.json
├── chromium-shared-sample-indexes-plate.csv
├── chromium-shared-sample-indexes-plate.json
├── chromium-single-cell-sample-indexes-plate-v1.csv
├── chromium-single-cell-sample-indexes-plate-v1.json
├── gemcode-single-cell-sample-indexes-plate.csv
└── gemcode-single-cell-sample-indexes-plate.json
10 directories, 34 files
Submetendo jobs
A execução do cellranger no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo cellranger.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=cellranger
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=16
#SBATCH --mem-per-cpu=4GB
module load cellranger
ID="sample1"
TRANSCRIPTOME="/public/cellranger/references/human/refdata-gex-GRCh38-2024-A"
FASTQS_DIR="/caminho/para/fastq_dir"
SAMPLE="SA001" # prefixo dos arquivos fastq
cellranger count --id="$ID" \
--transcriptome="$TRANSCRIPTOME" \
--fastqs="$FASTQS_DIR" \
--sample="$SAMPLE" \
--create-bam=true
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch cellranger.sh
Cellpose
O Cellpose é um modelo de segmentação de células baseado em aprendizado profundo, projetado para ser versátil e fácil de usar. Ele pode segmentar uma ampla variedade de tipos de células e tecidos sem a necessidade de treinamento específico para cada tipo.
Para mais informações sobre o Cellpose, acesse https://cellpose.org/.
Carregando o módulo
Para habilitar o Cellpose no HPCC Marvin, você deve carregar o módulo cellpose:
module load cellpose
As versões disponíveis do Cellpose no HPCC Marvin são:
cellpose/4.0.6 (D)cellpose/3.1.1.2cellpose/2.3.2
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help cellpose
Executando o Cellpose com interface gráfica (GUI)
A execução do Cellpose no HPCC Marvin é feita por meio de uma sessão VNC (Virtual Network Computing) utilizando o Open OnDemand. Para isso, siga os passos abaixo:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione a partição
gui-gpu-smalle defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o Cellpose:
# Habilitar o módulo
module load cellpose
# Iniciar o Cellpose com interface gráfica
cellpose
Submetendo jobs do Cellpose
O Cellpose também pode ser executado via submissão de jobs no SLURM, permitindo análises em segundo plano e melhor aproveitamento dos recursos do cluster. Crie um arquivo de script, por exemplo cellpose.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=cellpose
#SBATCH --partition=short-gpu-small
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=2GB
#SBATCH --gres=gpu:1g.5gb:1
module load cellpose
cellpose <parâmetros do cellpose>
Por exemplo, para segmentar imagens em uma pasta de entrada e salvar os resultados em uma pasta de saída, você pode usar:
cellpose --dir /home/carsen/images_cyto/test/ --save_png
Para mais detalhes sobre todos os parâmetros do Cellpose, use:
cellpose --help
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch cellpose.sh
Uso via Python (scripting)
O Cellpose também pode ser usado diretamente em scripts Python, permitindo automatizar análises e integrá-las a pipelines:
import numpy as np
import matplotlib.pyplot as plt
from cellpose import models, io
from cellpose.io import imread
io.logger_setup()
model = models.CellposeModel(gpu=True)
# list of files
# PUT PATH TO YOUR FILES HERE!
files = ['/media/carsen/DATA1/TIFFS/onechan.tif']
imgs = [imread(f) for f in files]
nimg = len(imgs)
masks, flows, styles = model.eval(imgs)
Cellprofiler
O CellProfiler é um programa de código aberto amplamente utilizado para análise de imagens biológicas. Ele permite a quantificação de características celulares em imagens, facilitando a análise de grandes conjuntos de dados.
Para mais informações sobre o CellProfiler, acesse https://cellprofiler.org/.
Carregando o módulo
Para habilitar o CellProfiler no HPCC Marvin, você deve carregar o módulo cellprofiler:
module load cellprofiler
As versões disponíveis do CellProfiler no HPCC Marvin são:
cellprofiler/4.2.6 (D)cellprofiler/4.2.4
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help cellprofiler
Executando o CellProfiler com interface gráfica (GUI)
A execução do CellProfiler no HPCC Marvin é feita por meio de uma sessão VNC (Virtual Network Computing) utilizando o Open OnDemand. Para isso, siga os passos abaixo:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione a partição
gui-gpu-smalle defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o CellProfiler:
# Habilitar o módulo
module load cellprofiler
# Iniciar o CellProfiler com interface gráfica
cellprofiler
Para conjuntos de dados muito grandes, recomenda-se utilizar a CLI do CellProfiler (sem interface gráfica) para obter melhor desempenho. O uso da GUI pode ser mais lento e consumir mais recursos, resultando em travamentos ou falhas na análise.
Submetendo jobs do CellProfiler
O CellProfiler também pode ser executado via submissão de jobs no SLURM, permitindo análises em segundo plano e melhor aproveitamento dos recursos do cluster. Crie um arquivo de script, por exemplo cellprofiler.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=cellprofiler
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=2GB
module load cellprofiler
cellprofiler -c -r -p /path/to/your/pipeline.cppipe -o /path/to/output -i /path/to/images
Os parâmetros utilizados nesse comando são:
-c: Executa o CellProfiler em modo CLI (sem interface gráfica).-r: Executa o pipeline na inicialização.-p: Especifica o caminho para o pipeline que será executado (pipeline.cppipe).-i: Caminho para a pasta com as imagens de entrada.-o: Caminho para a pasta onde os resultados serão salvos.
Caso utilize módulos que demandem GPU, selecione uma partição compatível:
short-gpu-small, adicione:#SBATCH --gres=gpu:a100:1short-gpu-big, adicione:#SBATCH --gres=gpu:1g.5gb:1
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch cellprofiler.sh
Para mais detalhes sobre os parâmetros do CellProfiler, use:
cellprofiler --help
Circos
O Circos é um pacote de software para visualização de dados e informações em um layout circular.
Ele é comumente utilizado para visualização de dados genômicos e criação de gráficos complexos, como:
- Rearranjos genômicos
- Conexões genômicas
- Mapas de calor (heatmaps) e gráficos de dispersão
- Relações de dados em layouts circulares
Para mais informações sobre o Circos, acesse: https://circos.ca/
Carregando o módulo
Para habilitar o Circos no HPCC Marvin, você deve carregar o módulo circos:
module load circos
As versões disponíveis do Circos no HPCC Marvin são:
circos/0.69-10 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help circos
Configurando o módulo
O Circos gera imagens estáticas e o processo de geração das imagens é gerenciado por um arquivo de configurações central. Esse arquivo geralmente importa outros arquivos de configurações, como preferências de fonte e cores.
Para executar o Circos, é necessário passar como argumento um arquivo de configuração com a flag
-conf [conf_file].
Acesse Circos: Configuration files para mais informações sobre sintaxe dos arquivos de configuração e como organizar os blocos.
Durante a execução, caso não seja explicitamente definido o arquivo de configuração, o Circos buscará automaticamente por um circos.conf nos seguintes caminhos (entre outros):
./circos.conf
./etc/circos.conf
./../etc/circos.conf
Acesse Circos: Runtime parameters para mais informações.
O módulo contém o diretório example que pode ser utilizado como referência e para testar a execução do mesmo.
Você pode copiar o diretório para o seu home e executar o circos a partir dele, por exemplo:
cd ~
cp -r /opt/images/apps/circos/v0.69-10/example .
cd example
circos
Submetendo jobs
A execução do Circos no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo circos.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=circos
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=2GB
module load circos/0.69-10
CONFIG_FILE="/caminho/para/circos.conf"
circos -conf "$CONFIG_FILE"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch circos.sh
Para mais detalhes sobre os parâmetros do Circos, use:
circos --help
cisTEM
O cisTEM (Computational Imaging System for Transmission Electron Microscopy) é um software para o processamento de imagens de criomicroscopia eletrônica (cryo-EM) de complexos macromoleculares, permitindo a obtenção de reconstruções 3D em alta resolução. O software reúne diversas ferramentas para processamento de dados de imagens — incluindo filmes, micrografias e pilhas de partículas únicas — oferecendo um pipeline completo para reconstruções de partículas únicas em alta resolução.
Para mais informações sobre o cisTEM, acesse https://cistem.org/documentation.
Carregando o módulo
O cisTEM está disponível dentro do módulo scipion/3.8.3. Para utilizá-lo no HPCC Marvin, você deve carregar o módulo scipion/3.8.3:
module load scipion/3.8.3
Para acessar a documentação do modulo, utilize:
module help scipion/3.8.3
Como executar o cisTEM no Open OnDemand
Para executar o cisTEM, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o cisTEM:
# Habilitar o módulo
module load scipion/3.8.3
# Iniciar o cisTEM com interface gráfica
cistem
Cooler
O Cooler é uma ferramenta e biblioteca Python para armazenar, manipular e acessar matrizes de contato Hi-C de forma eficiente.
Ele utiliza o formato .cool, baseado em HDF5, que permite consultas rápidas e armazenamento compacto de dados de interação cromossômica.
O Cooler fornece tanto utilitários de linha de comando quanto interfaces Python, possibilitando a criação, indexação e análise de dados Hi-C em diferentes resoluções.
Para mais informações, acesse: https://cooler.readthedocs.io/en/stable/
Carregando o módulo
Para habilitar o cooler no HPCC Marvin, você deve carregar o módulo cooler:
module load cooler
As versões disponíveis do cooler no HPCC Marvin são:
cooler/0.10.4 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help cooler
Executando o módulo
O pacote cooler incluí diversos comandos que são utilizados no para criar, realizar queries e manipular arquivos cooler. Modelo de uso do cooler via linha de comando:
cooler [OPTIONS] COMMAND [ARGS]...
Ao executar cooler -h, é possível consultar a lista de comandos disponíveis:
Commands:
balance Out-of-core matrix balancing.
cload Create a cooler from genomic pairs and bins.
coarsen Coarsen a cooler to a lower resolution.
csort Sort and index a contact list.
digest Generate fragment-delimited genomic bins.
dump Dump a cooler's data to a text stream.
ls List all coolers inside a file.
cp Copy a cooler from one file to another or within the same file.
ln Create a hard link to a cooler (rather than a true copy) in...
mv Rename a cooler within the same file.
tree Display a file's data hierarchy.
attrs Display a file's attribute hierarchy.
info Display a cooler's info and metadata.
load Create a cooler from a pre-binned matrix.
makebins Generate fixed-width genomic bins.
merge Merge multiple coolers with identical axes.
show Display and browse a cooler in matplotlib.
zoomify Generate a multi-resolution cooler file by coarsening.
Consulte todos informações completas sobre os comandos na página de referência de CLI na documentação oficial do cooler.
Submetendo jobs
A execução do cooler no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo cooler.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=cooler
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=4GB
module load cooler/0.10.4
BINS="/caminho/para/bed/file"
PAIRS_PATH="/caminho/para/contacts/file"
COOL_PATH="/caminho/para/output/cool/file"
cooler cload pairs "$BINS" "$PAIRS_PATH" "$COOL_PATH"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch cooler.sh
Para mais detalhes sobre os parâmetros de cada comando do cooler, use:
cooler [COMMAND] -h
cooltools
O cooltools é uma biblioteca Python voltada para a análise avançada de dados Hi-C. Ela se integra diretamente com arquivos .cool, oferecendo funções para calcular métricas de interação cromossômica, como isolamento de domínios TAD, correlações de compartimentos, métricas de cis/trans e muito mais. O cooltools combina utilitários de linha de comando e funções Python, permitindo análises flexíveis e reprodutíveis em diferentes resoluções de dados Hi-C.
Para mais informações e documentação completa, acesse: https://cooltools.readthedocs.io/en/latest/
Carregando o módulo
Para habilitar o cooltools no HPCC Marvin, você deve carregar o módulo cooltools:
module load cooltools
As versões disponíveis do cooltools no HPCC Marvin são:
cooltools/0.7.1 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help cooltools
Executando o módulo
Modelo de uso do cooltools via linha de comando:
cooltools [OPTIONS] COMMAND [ARGS]...
Ao executar cooltools -h, é possível consultar a lista de comandos disponíveis:
Commands:
coverage Calculate the sums of cis and genome-wide contacts (aka...
dots Call dots on a Hi-C heatmap that are not larger than...
eigs-cis Perform eigen value decomposition on a cooler matrix to...
eigs-trans Perform eigen value decomposition on a cooler matrix to...
expected-cis Calculate expected Hi-C signal for cis regions of...
expected-trans Calculate expected Hi-C signal for trans regions of...
genome Utilities for binned genome assemblies.
insulation Calculate the diamond insulation scores and call...
pileup Perform retrieval of the snippets from .cool file.
random-sample Pick a random sample of contacts from a Hi-C map.
rearrange Rearrange data from a cooler according to a new genomic...
saddle Calculate saddle statistics and generate saddle plots...
virtual4c Generate virtual 4C profile from a contact map by...
Consulte todos informações completas sobre os comandos na página de referência de CLI na documentação oficial do cooltools.
Submetendo jobs
A execução do cooltools no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo cooltools.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=cooltools
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=4GB
module load cooltools/0.7.1
COOL_PATH="/caminho/para/arquivo.cool"
OUTPUT="/caminho/para/output.tsv"
cooltools coverage -o "$OUTPUT" "$COOL_PATH"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch cooltools.sh
Para mais detalhes sobre os parâmetros de cada comando do cooltools, use:
cooltools [COMMAND] -h
deepTools
O deepTools é um conjunto de ferramentas para análise e visualização de dados de sequenciamento de próxima geração (NGS), especialmente voltado para experimentos baseados em alinhamentos como ChIP-seq, RNA-seq e ATAC-seq.
Ele oferece utilitários para normalizar, comparar e representar graficamente dados em formato BAM, bigWig ou bedGraph, permitindo análises detalhadas de cobertura genômica, perfis de enriquecimento e padrões epigenéticos.
O deepTools é amplamente utilizado em bioinformática e genômica funcional para explorar relações entre marcas epigenéticas, expressão gênica e estrutura cromatínica.
Para mais informações, acesse: https://deeptools.readthedocs.io/en/latest/index.html
Carregando o módulo
Para habilitar o deeptools no HPCC Marvin, você deve carregar o módulo deeptools:
module load deeptools
As versões disponíveis do deeptools no HPCC Marvin são:
deeptools/3.5.6 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help deeptools
Executando o módulo
O deeptools consiste em diversas ferramentas que podem ser chamadas pelo próprio nome, por exemplo:
multiBamSummary bins --bamfiles file1.bam file2.bam -o results.npz
Algumas outras ferramentas são:
findRestSite Identifies the genomic locations of restriction sites
hicBuildMatrix Creates a Hi-C matrix using the aligned BAM files of the Hi-C sequencing reads
hicQuickQC Estimates the quality of Hi-C dataset
hicQC Plots QC measures from the output of hicBuildMatrix
hicCorrectMatrix Uses iterative correction to remove biases from a Hi-C matrix
Consulte todas as ferramentas disponíveis com deeptools -h e mais informações na documentação oficial do deeptools.
Submetendo jobs
A execução do deeptools no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo deeptools.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=deeptools
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem-per-cpu=4GB
module load deeptools/3.5.6
BAM_FILE_1="/caminho/para/bamfile1"
BAM_FILE_2="/caminho/para/bamfile2"
OUTPUT_FILE="results.npz"
multiBamSummary bins --bamfiles "$BAM_FILE_1" "$BAM_FILE_2" -o "$OUTPUT_FILE"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch deeptools.sh
Para mais detalhes sobre os parâmetros do deeptools, use:
deeptools -h
Etomo
O Etomo é um software para a reconstrução de tomografias eletrônicas, permitindo a visualização e análise de dados de tomografia em 3D. O Etomo é parte do pacote IMOD e é amplamente utilizado na comunidade de microscopia eletrônica.
Carregando o módulo
O Etomo está disponível dentro do módulo scipion/3.8.3. Para utilizá-lo no HPCC Marvin, você deve carregar o módulo scipion/3.8.3:
module load scipion/3.8.3
Para acessar a documentação do modulo, utilize:
module help scipion/3.8.3
Como executar o Etomo no Open OnDemand
Para executar o Etomo, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o Etomo:
# Habilitar o módulo
module load scipion/3.8.3
# Iniciar o Etomo com interface gráfica
etomo
Para mais detalhes sobre os parâmetros do Etomo, use:
etomo --help
FastQC
O FastQC é um software para análise de controle de qualidade de dados de sequenciamento (NGS – Next Generation Sequencing).
Ele é amplamente utilizado para avaliar a qualidade de arquivos FASTQ, fornecendo uma visão geral rápida e detalhada sobre os dados antes de etapas posteriores de análise.
O FastQC gera relatórios gráficos e estatísticos que permitem identificar possíveis problemas nas leituras, como:
- Qualidade das bases ao longo das sequências
- Conteúdo GC e distribuição de tamanhos de leitura
- Presença de adaptadores ou contaminantes
- Sequências duplicadas
- Overrepresented sequences (sequências com frequência anormalmente alta)
Para mais informações sobre o FastQC, acesse:
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Carregando o módulo
Para habilitar o fastqc no HPCC Marvin, você deve carregar o módulo fastqc:
module load fastqc
As versões disponíveis do fastqc no HPCC Marvin são:
fastqc/0.12.1 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help fastqc
Como executar o fastqc no Open OnDemand
Para executar o fastqc, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-cpu,gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o fastqc:
# Habilitar o módulo
module load fastqc/0.12.1
# Iniciar o fastqc com interface gráfica
fastqc
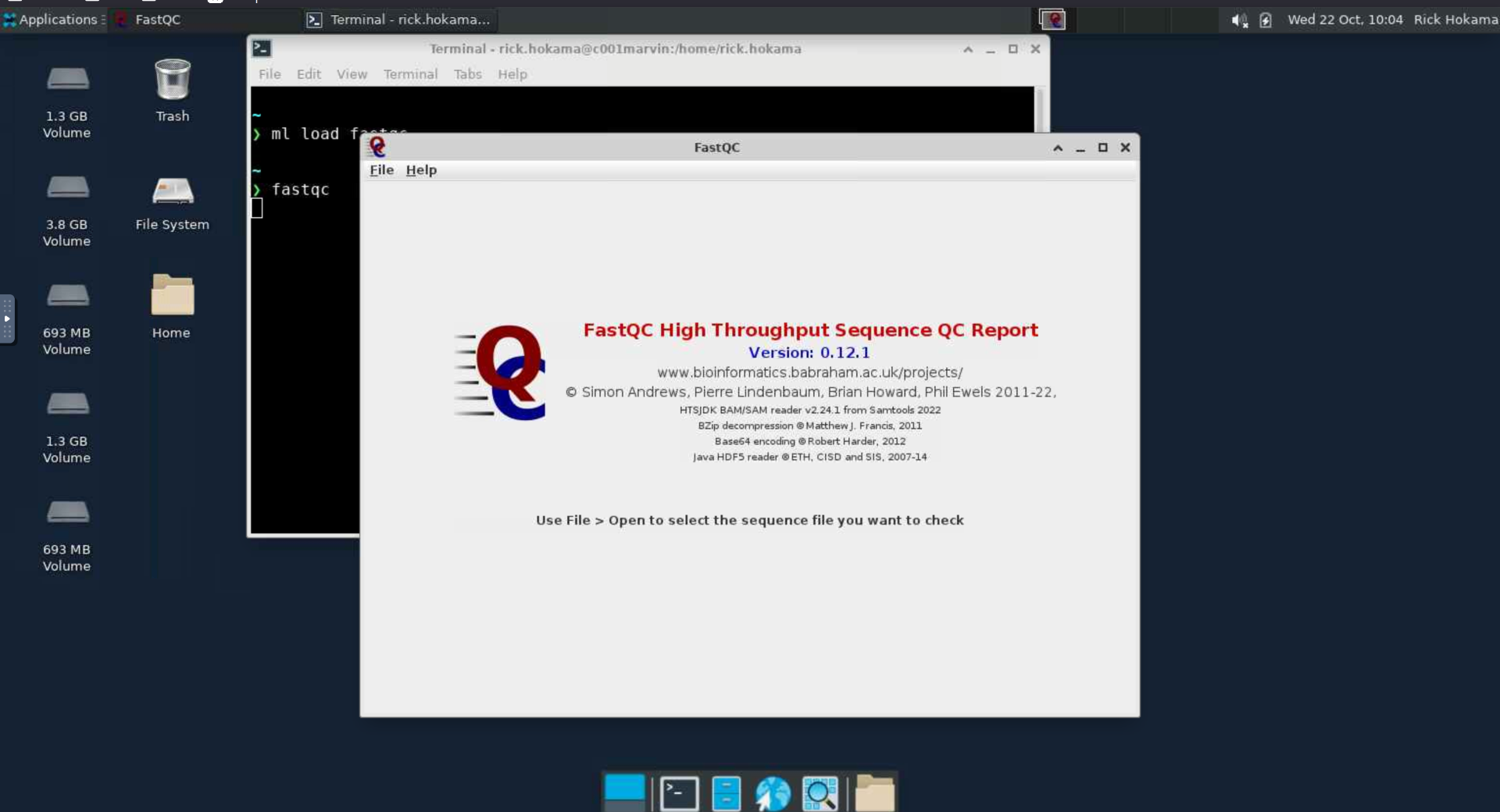
Para mais detalhes sobre os parâmetros do Fastqc, use:
fastqc --help
Como executar o fastqc via SSH
Ao conectar remotamente no Marvin via SSH, é necessário habilitar o X11 forwarding para exibir a interface gráfica na máquina local.
# Iniciar conexão ssh habilitando X11 forwarding
ssh -X <user>@marvin.cnpem.br
# Habilitar módulo
module load fastqc/0.12.1
# Iniciar o fastqc com interface gráfica
fastqc
Fiji
O Fiji (Fiji Is Just ImageJ) é uma distribuição do ImageJ, um software de código aberto amplamente utilizado para análise e processamento de imagens científicas. O Fiji inclui uma série de plugins e ferramentas adicionais que facilitam tarefas comuns em análise de imagens biológicas.
Para mais informações sobre o Fiji, acesse https://fiji.sc/.
Carregando o módulo
Para habilitar o Fiji no HPCC Marvin, você deve carregar o módulo fiji:
module load fiji
As versões disponíveis do Fiji no HPCC Marvin são:
fiji/2.16.0
Para acessar a documentação do modulo, utilize:
module help fiji
Executando o Fiji com interface gráfica (GUI)
A execução do Fiji no HPCC Marvin é feita por meio de uma sessão VNC (Virtual Network Computing) utilizando o Open OnDemand. Para isso, siga os passos abaixo:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione a partição
gui-gpu-smalle defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o Fiji:
# Habilitar o módulo
module load fiji
# Iniciar o Fiji com interface gráfica
fiji
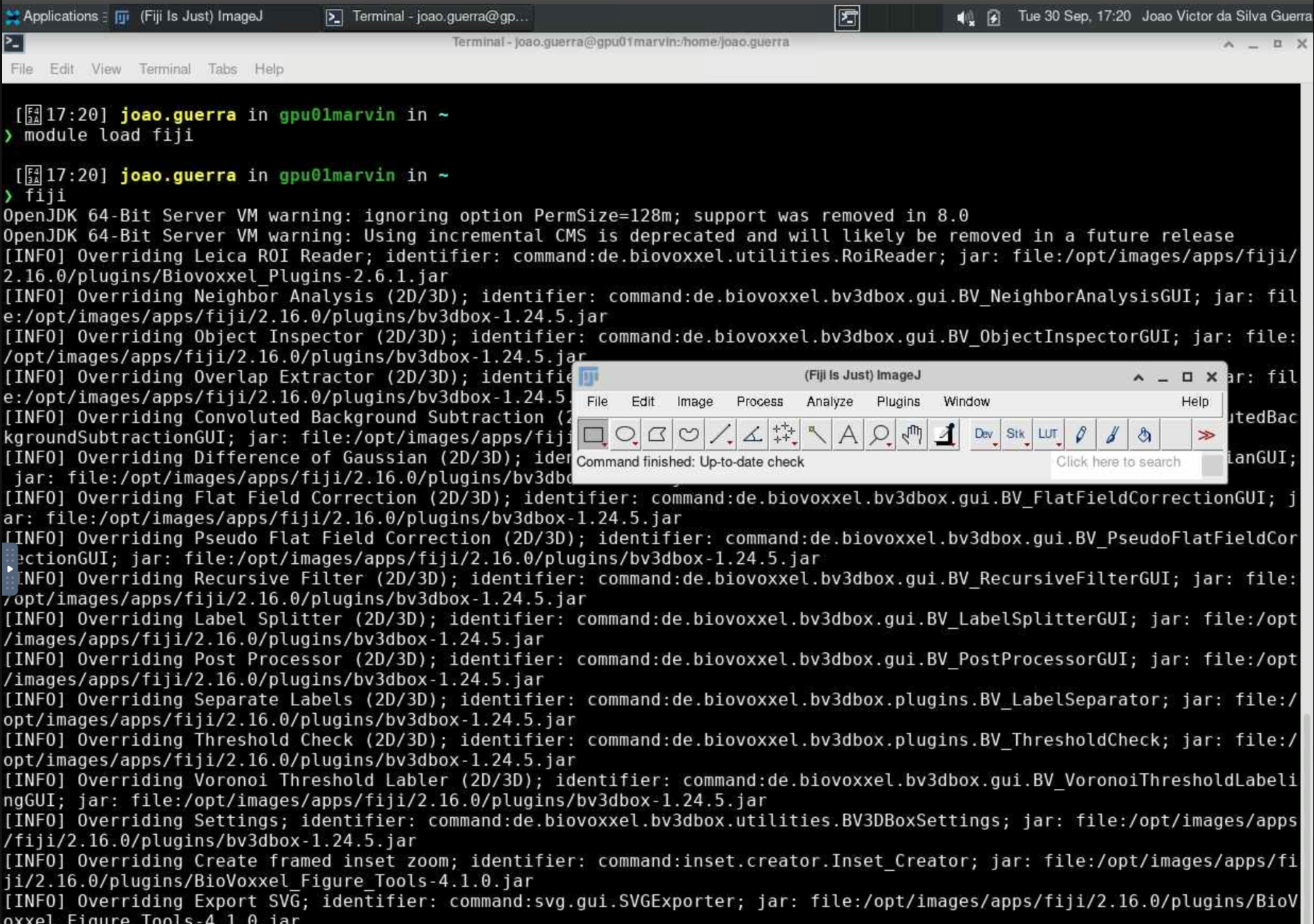
Para mais detalhes sobre os parâmetros do Fiji, use:
fiji --help
GTDB-Tk
O GTDB-Tk (Genome Taxonomy Database Toolkit) é uma ferramenta desenvolvida para classificar genomas bacterianos e arqueanos de forma padronizada com base no Genome Taxonomy Database (GTDB). Ele utiliza abordagens filogenéticas e de similaridade genômica (ANI) para atribuir classificações taxonômicas consistentes e atualizadas, garantindo maior precisão e reprodutibilidade nas análises de genomas microbianos.
O GTDB-Tk é amplamente utilizado em estudos de metagenômica e genômica comparativa, integrando-se facilmente a pipelines bioinformáticos para análise em larga escala.
Para mais informações e documentação completa, acesse: https://ecogenomics.github.io/GTDBTk/.
Carregando o módulo
Para habilitar o GTDB-Tk no HPCC Marvin, você deve carregar o módulo gtdbtk:
module load gtdbtk
As versões disponíveis do gtdbtk no HPCC Marvin são:
gtdbtk/2.5.2 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help gtdbtk
Dados de referência
Para executar o GTDB-Tk, é necessário o download da database com dados de referência, que pesa em torno de 140G.
Atualmente, a database de referência está localizada em /public/gtdbtk_reference_data/.
Caso seja necessário alterar o local dos dados de referência que desejar utilizar, é necessário configurar a variável GTDBTK_DATA_PATH antes de executar o módulo:
export GTDBTK_DATA_PATH="/novo/path/desejado/do/reference/data/"
Executando o módulo
Modelo de uso do GTDB-Tk via linha de comando:
gtdbtk COMMAND [ARGS]...
Ao executar gtdbtk -h, é possível consultar a lista de comandos disponíveis:
Workflows:
classify_wf -> Classify genomes by placement in GTDB reference tree
(ani_screening -> identify -> align -> classify)
de_novo_wf -> Infer de novo tree and decorate with GTDB taxonomy
(identify -> align -> infer -> root -> decorate)
Methods:
identify -> Identify marker genes in genome
align -> Create multiple sequence alignment
classify -> Determine taxonomic classification of genomes
infer -> Infer tree from multiple sequence alignment
root -> Root tree using an outgroup
decorate -> Decorate tree with GTDB taxonomy
Tools:
infer_ranks -> Establish taxonomic ranks of internal nodes using RED
ani_rep -> Calculates ANI to GTDB representative genomes
trim_msa -> Trim an untrimmed MSA file based on a mask
export_msa -> Export the untrimmed archaeal or bacterial MSA file
remove_labels -> Remove labels (bootstrap values, node labels) from an Newick tree
convert_to_itol -> Convert a GTDB-Tk Newick tree to an iTOL tree
convert_to_species -> Convert GTDB genome IDs to GTDB species names
Testing:
test -> Validate the classify_wf pipeline with 3 archaeal genomes
check_install -> Verify third party programs and GTDB reference package
Consulte todos informações completas sobre os comandos na página de referência de comandos na documentação oficial do gtdbtk.
Submetendo jobs
A execução do GTDB-Tk no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo gtdbtk.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=gtdbtk
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=32
#SBATCH --mem-per-cpu=8GB
module load gtdbtk/2.5.2
GENOME_DIR="/caminho/para/diretorio/genoma"
OUTPUT_DIR="/caminho/para/diretorio/output"
gtdbtk classify_wf --genome-dir_ ${GENOME_DIR} --out_dir ${OUTPUT_DIR}
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch gtdbtk.sh
Para mais detalhes sobre os parâmetros de cada comando do GTDB-Tk, use:
gtdbtk [COMMAND] -h
GROMACS
O GROMACS (GROningen MAchine for Chemical Simulations) é um conjunto de softwares livres e de código aberto para simulação e análise de dinâmica molecular. Ele é amplamente utilizado para o estudo de sistemas biológicos, como proteínas, lipídios e ácidos nucleicos, mas também pode ser aplicado a sistemas não biológicos.
Para mais informações sobre o GROMACS, acesse https://manual.gromacs.org/current/index.html/.
Carregando o módulo
Para habilitar o GROMACS no HPCC Marvin, você deve carregar o módulo gromacs:
module load gromacs
As versões disponíveis do GROMACS no HPCC Marvin são:
gromacs/2024.5 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help gromacs
Submetendo jobs
A execução do GROMACS no HPCC Marvin é feita por meio de scripts de submissão no SLURM.
Por padrão, recomenda-se utilizar a fila short-gpu-small para execuções de pequeno e médio porte. Para isso, crie um arquivo de script, por exemplo gromacs.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=gromacs
#SBATCH --partition=short-gpu-small
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem-per-cpu=2GB
#SBATCH --gres=gpu:1g.5gb:1
#SBATCH --time=24:00:00
# Load GROMACS module
module load gromacs/2024.5
# Run molecular dynamics simulation with GPU acceleration
gmx mdrun -s production.tpr -v -deffnm production -pin off -ntomp $SLURM_CPUS_PER_TASK -nb gpu -pme gpu -update gpu -bonded gpu
Para execuções de maior porte, utilize a fila short-gpu-big e ajuste os parâmetros de recursos conforme o exemplo abaixo:
#!/bin/bash
#SBATCH --job-name=gromacs
#SBATCH --partition=short-gpu-big
#SBATCH --ntasks=3
#SBATCH --cpus-per-task=8
#SBATCH --mem-per-cpu=2GB
#SBATCH --gres=gpu:a100:1
#SBATCH --time=24:00:00
# Load GROMACS module
module load gromacs/2024.5
# Run molecular dynamics simulation with GPU acceleration
gmx mdrun -s production.tpr -v -deffnm production -pin off -ntomp $SLURM_CPUS_PER_TASK -nb gpu -pme gpu -update gpu -bonded gpu
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch gromacs.sh
Para mais detalhes sobre os comandos do GROMACS, use:
gmx help
hic2cool
O hic2cool é uma ferramenta leve para converter matrizes de contato Hi-C do formato .hic para o formato .cool.
Ele suporta arquivos de resolução única ou múltiplas resoluções, permitindo integrar facilmente dados Hi-C com outras ferramentas de análise e visualização.
O hic2cool pode ser utilizado tanto como pacote Python em scripts quanto como ferramenta de linha de comando, oferecendo flexibilidade para pipelines automatizados de processamento de dados Hi-C.
Para mais informações, acesse: https://github.com/4dn-dcic/hic2cool/
Carregando o módulo
Para habilitar o hic2cool no HPCC Marvin, você deve carregar o módulo hic2cool:
module load hic2cool
As versões disponíveis do hic2cool no HPCC Marvin são:
hic2cool/1.0.1 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help hic2cool
Executando o módulo
O hic2cool possui diferentes modos que devem ser selecionados ao executá-lo. Segue um exemplo de convert:
hic2cool convert <infile> <outfile> -r <resolution> -p <nproc>
Consulte todos os modos de execução disponíveis com hic2cool -h e mais informações na no repositório do hi2cool.
Submetendo jobs
A execução do hic2cool no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo hic2cool.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=hic2cool
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=2GB
module load hic2cool/1.0.1
INPUT_FILE="/caminho/para/input/file"
OUTPUT_FILE="/caminho/para/input/file"
RESOLUTION=0
NPROC=1
hic2cool convert "$INPUT_FILE" "$OUTPUT_FILE" -r "$RESOLUTION" -p "$NPROC"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch hic2cool.sh
Para mais detalhes sobre os parâmetros do hic2cool, use:
hic2cool -h
HiCExplorer
O HiCExplorer é um conjunto de ferramentas para análise, processamento e visualização de dados de experimentos Hi-C, que investigam a organização tridimensional do genoma.
Ele permite construir e normalizar matrizes de contato, corrigir vieses técnicos, identificar domínios topológicos (TADs) e gerar visualizações de alta qualidade, como mapas de calor e comparações entre amostras.
O HiCExplorer é amplamente utilizado em estudos de arquitetura genômica, regulação gênica e epigenética.
Para mais informações, acesse: https://hicexplorer.readthedocs.io/en/latest/
Carregando o módulo
Para habilitar o hicexplorer no HPCC Marvin, você deve carregar o módulo hicexplorer:
module load hicexplorer
As versões disponíveis do hicexplorer no HPCC Marvin são:
hicexplorer/3.7.6 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help hicexplorer
Executando o módulo
O HiCExplorer consiste em diversas ferramentas que podem ser chamadas pelo próprio nome, por exemplo:
hicPlotMatrix -m hic_matrix.h5 -o plot.pdf
Algumas outras ferramentas são:
findRestSite Identifies the genomic locations of restriction sites
hicBuildMatrix Creates a Hi-C matrix using the aligned BAM files of the Hi-C sequencing reads
hicQuickQC Estimates the quality of Hi-C dataset
hicQC Plots QC measures from the output of hicBuildMatrix
hicCorrectMatrix Uses iterative correction to remove biases from a Hi-C matrix
Consulte todas as ferramentas disponíveis com hicexplorer -h e mais informações na documentação oficial do hicexplorer.
Submetendo jobs
A execução do HiCExplorer no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo hicexplorer.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=hicexplorer
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem-per-cpu=4GB
module load hicexplorer/3.7.6
INPUT_MATRIX="/caminho/para/matriz"
OUTPUT="/caminho/para/output/desejado"
hicPlotMatrix -m "$INPUTMATRIX" -o "$OUTPUT"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch hicexplorer.sh
Para mais detalhes sobre os parâmetros do HiCExplorer, use:
hicexplorer -h
HiCsuntdracones
O HiC-sunt-dracones é um software para análise e comparação de dados Hi-C em larga escala.
Ele foi desenvolvido para identificar diferenças estruturais na organização tridimensional do genoma entre condições ou tipos celulares distintos.
A ferramenta realiza o pré-processamento, normalização e detecção de regiões diferencialmente organizadas (DORs), permitindo avaliar alterações em domínios topológicos (TADs) e padrões de interação cromossômica.
O HiC-sunt-dracones é amplamente utilizado em estudos de epigenômica e arquitetura nuclear, fornecendo uma abordagem estatística robusta para comparar dados Hi-C entre múltiplas amostras.
Para mais informações, acesse: https://github.com/ComputationalEpigenetics/hic-sunt-dracones
Carregando o módulo
Para habilitar o hicsuntdracones no HPCC Marvin, você deve carregar o módulo hicsuntdracones:
module load hicsuntdracones
As versões disponíveis do hicsuntdracones no HPCC Marvin são:
hicsuntdracones/0.2.0 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help hicsuntdracones
Executando o módulo
Após carregar o hicsuntdracones, ele pode ser executado tanto com sua abreviação hicsd, como com seu nome completo hicsuntdracones.
O consiste em diversos comandos, por exemplo:
hicsd hicpro2homer [-h] --input_bed INPUT_BED --input_matrix INPUT_MATRIX --output_matrix OUTPUT_MATRIX
Lista completa dos comandos:
version Show version
hicpro2homer Convert a matrix in HiC-Pro format to Homer format
mHiC2homer Convert a matrix in mHiC format to Homer format
number_of_bins Return the number of bins of the matrix
chromosomes Return the chromosomes used in the matrix
norm_by_col_sum Normalize by column sum to make matrices comparable
submatrix Extract submatrices
diff_matrix Generate differential matrix by dividing the values of one matrix by the values of the other.
heatmap Plot interaction matix heatmap
histogram Plot histogram for matrix reads
virtual_4C Perform virtual 4C analysis
dist_dep_decay Distant dependent decay
colo Perform colocalisation analysis
colo_diff Perform differential colocalisation analysis
ploidy Apply ploidy factors
Consulte todas as ferramentas disponíveis com hicsd -h.
Submetendo jobs
A execução do hicsuntdracones no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo hicsuntdracones.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=hicsuntdracones
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem-per-cpu=4GB
module load hicsuntdracones/0.2.0
INPUT_BED="/caminho/para/input/bed"
INPUT_MATRIX="/caminho/para/input/matrix"
OUTPUT_MATRIX="/caminho/para/output/matrix"
hicsd hicpro2homer [-h] --input_bed INPUT_BED --input_matrix INPUT_MATRIX --output_matrix OUTPUT_MATRIX
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch hicsuntdracones.sh
Para mais detalhes sobre os parâmetros do HiCsuntdracones, use:
hicsuntdracones -h
Ilastik
O Ilastik é uma ferramenta de aprendizado de máquina interativa para análise de imagens, especialmente útil para tarefas como segmentação, classificação e rastreamento de objetos em imagens biológicas.
Para mais informações sobre o Ilastik, acesse https://www.ilastik.org/documentation/.
Carregando o módulo
Para habilitar o Ilastik no HPCC Marvin, você deve carregar o módulo ilastik:
module load ilastik
As versões disponíveis do Ilastik no HPCC Marvin são:
ilastik/1.4.1 (D)ilastik/1.4.0
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help ilastik
Como executar o Ilastik no Open OnDemand
Para executar o Ilastik, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione a partição
gui-gpu-smalle defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o Ilastik:
# Habilitar o módulo
module load ilastik
# Iniciar o Ilastik com interface gráfica
ilastik
Submetendo jobs do Ilastik
O Ilastik também pode ser executado via submissão de jobs no SLURM, permitindo análises em segundo plano e melhor aproveitamento dos recursos do cluster. Crie um arquivo de script, por exemplo ilastik.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=ilastik
#SBATCH --partition=short-gpu-small
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=2GB
#SBATCH --gres=gpu:1g.5gb:1
module load ilastik
# Executar o Ilastik em modo headless (sem interface gráfica)
ilastik --headless --project proj.ilp
Os parâmetros utilizados nesse comando são:
--headless: Executa o Ilastik em modo CLI (sem interface gráfica).--project proj.ilp: Especifica o caminho para o projeto que será executado.
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch ilastik.sh
Para mais detalhes sobre os parâmetros do Ilastik, use:
ilastik --help
IsoNet
O IsoNet (Isotropic Reconstruction For Electron Tomography) é uma ferramenta baseada em aprendizado profundo desenvolvida para melhorar a reconstrução de volumes em tomografia eletrônica.
Para mais informações sobre o IsoNet, acesse https://isonetcryoet.com/docs.html. O código-fonte está disponível em https://github.com/IsoNet-cryoET/IsoNet.
Carregando o módulo
O IsoNet está disponível dentro do módulo scipion/3.8.3. Para utilizá-lo no HPCC Marvin, você deve carregar o módulo scipion/3.8.3:
module load scipion/3.8.3
Para acessar a documentação do modulo, utilize:
module help scipion/3.8.3
Como executar o IsoNet no Open OnDemand
Para executar o IsoNet, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o IsoNet:
# Habilitar o módulo
module load scipion/3.8.3
# Iniciar o IsoNet
isonet
# Iniciar o IsoNet com interface gráfica
isonet gui
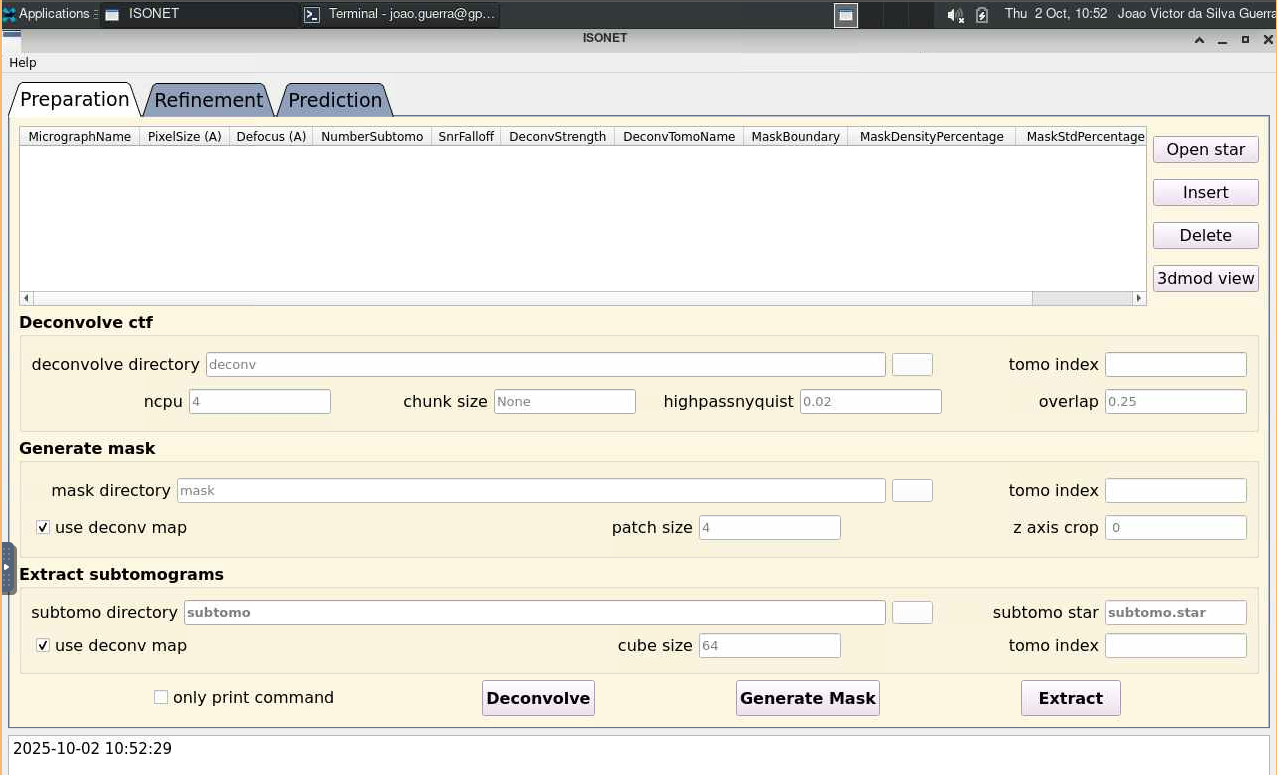
Para mais detalhes sobre os parâmetros do IsoNet, use:
isonet --help
Juicer
Juicer é um pipeline completo para processamento e análise de dados Hi-C, desenvolvido pelo Aiden Lab. Ele automatiza desde o alinhamento das leituras até a geração de mapas de interação genômica em múltiplas resoluções (arquivos .hic).
Útil para explorar estruturas 3D do genoma — como TADs, loops e compartimentos — o Juicer é amplamente adotado em genômica estrutural.
Para mais informações, acesse https://github.com/aidenlab/juicer/wiki.
Carregando o módulo
Para habilitar o juicer no HPCC Marvin, você deve carregar o módulo juicer:
module load juicer/<versão desejada>
As versões disponíveis do juicer no HPCC Marvin são:
juicer/2.0 (D)juicer/1.6
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help juicer
Executando o módulo
O script principal do juicer é chamado com o comando juicer.sh e pode ser executado conforme o exemplo:
juicer.sh -z [reference genome file]
Para mais informações detalhadas sobre o script do juicer, acesse a página de Usage do juicer.
Além do script principal, o módulo também conta com o pacote de ferramentas juicer_tools. Para obter informações sobre o uso e ajuda, execute:
juicer_tools -h
Para mais informações, acesse a página de Quick Start do juicer tools.
Note que, na wiki do juicer tools, ele estará referenciado como juicebox_tools.jar e chama o java para executar o .jar. Isso não é necessário no Marvin, apenas passe as opções e comandos diretamente.
Por exemplo, caso você queira executar o seguinte comando que está na wiki do juicer tools:
java -Xmx2g -jar juicebox_tools.jar pre data/test.txt.gz data/test.hic hg19
No Marvin, você executaria da seguinte forma:
juicer_tools pre data/test.txt.gz data/test.hic hg19
Submetendo jobs
A execução do juicer no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo juicer.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=juicer
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem-per-cpu=4GB
module load juicer/2.0
REFERENCE_GENOME="caminho/para/genome/file"
juicer.sh -z "$REFERENCE_GENOME"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch juicer.sh
Para mais detalhes sobre os parâmetros do juicer, use:
juicer.sh -h
Kraken2
O Kraken2 é uma ferramenta de classificação taxonômica ultrarrápida para dados de sequenciamento metagenômico, baseada na correspondência exata de k-mers.
Ele permite identificar rapidamente a composição taxonômica de amostras biológicas a partir de leituras de DNA, atribuindo cada sequência ao nível taxonômico mais específico possível.
O Kraken2 é amplamente utilizado em estudos de microbiomas, viromas e análises ambientais, oferecendo alto desempenho e precisão por meio de índices otimizados e bases de dados personalizáveis.
Para mais informações, acesse: https://github.com/DerrickWood/kraken2/
Carregando o módulo
Para habilitar o kraken2 no HPCC Marvin, você deve carregar o módulo kraken2:
module load kraken2
As versões disponíveis do kraken2 no HPCC Marvin são:
kraken2/2.1.6 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help kraken2
Executando o módulo
Na segunda versão do kraken, é possível executá-lo via um wrapper script com o binário k2 em combinação com alguns comandos como add-to-library e build. O k2 oferece suporte a todas as opções de linha de comando dos scripts originais e adiciona alguns novos recursos que estão documentados no manual.
Por exemplo, para verificar obter o help do comando build, execute:
k2 build -h
Consulte todos os comandos disponíveis com k2 -h e documentação mais detalhada no manual do Kraken2 no GitHub.
Submetendo jobs
A execução do kraken2 no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo kraken2.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=kraken2
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem-per-cpu=6GB
module load kraken2/2.1.6
DB_PATH="/caminho/para/kraken.db"
k2 build --db "$DB_PATH"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch kraken2.sh
Para mais detalhes sobre os parâmetros do kraken2, use:
k2 -h
NP³ MS Workflow - A Pipeline for LC-MS/MS Metabolomics Data Process and Analysis
O NP³ MS Workflow (v1.1.4) é uma coleção de scripts para melhorar agilizar a pesquisa em metabolômica untarget focada em descoberta de drogas com otimizações para produtos naturais.
É um procedimento automatizado para agrupar e quantificar os espectros MS² (MS/MS) associadas ao mesmo íon que foram eluídos em picos cromatográficos (MS1) concorrentes em uma coleção de amostras de experimentos LC-MS/MS.
Os possíveis resultados são:
- Tabela classificação de espectros candidatos responsáveis por hits observados em experimentos de bioatividade;
- Quantificação relativa de metabólitos presentes nas amostras;
- Grafo de redes moleculares para melhorar a análise e visualização dos resultados.
Tutorial em vídeo
- Tutorial 1: Brief introduction, ‘Getting Started’ section and a [M+H]+ analysis
Para entender melhor
Criando arquivo sbatch
Para usar o NP³ MS Workflow no HPC MARVIN, a forma mais eficiente é submentendo um job no seu gerenciador de filas slurm.
Neste job colocamos todos os comandos que devem ser executados em um arquivo de texto simples de extesão .sh.
Cabeçalho
No cabeçalho os principais parametros são:
--jobname=<jobname>| Nome do seu job;--ntaks=<numero>| Número tarefas, geralmente 1 é suficiente;--cpus-per-task=<numero>| Número de CPUS que serão usadas, se usar um valor diferente de1lembre-se também modificar o parâmetro-l(é a letra éle) em seurun;--partition=<nome_particao>| O NP³ não é otimizado para GPU, então escolha uma das filas de CPU (que geralmente são menos concorridas, então pode ser uma vantagem). As fila de CPU atualmente disponíveis são:- debug-cpu | até 30:00 min | Recomendada para testes;
- gui-cpu | até 12:00:00 horas | Recomendada para sessões VNC no OOD;
- short-cpu | até 5-00:00:00 dias | Recomendada para maioria dos trabalhos;
- long-cpu | até 15-00:00:0 dias | Recomendada para processamentos muito demorados.
Exemplo de cabeçalho:
#!/bin/bash
#SBATCH --job-name=NP3_MS_WORKFLOW_your_intuitive_name
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --partition=short-cpu
Corpo
No corpo do arquivo colocamos os comandos que serão executados. Nesse caso, é o comando para executar o np3_ms_workflow instalado dentro de um container do singularity e os parâmetros para execução.
Execução do NP³
Depois de ter enviado seus arquivos MZXML e metadado à sua área do OpenOnDemand, basta construir seu comando de processamento utilizando os endereços completos dos diretórios
Abaixo um exemplo para o run:
- Da usuário
marie.curie; - Saída no diretório
tmpsdentro de suahome; - Com metadao em pasta de mzxml em
Documentos\NPTest, também dentro dahome; - Tempos de tolerância de 1s para o mesmo batch e 2s geral;
- Verbose 10 que significa ligado para registro e ativa o teste de integridade no final (consulte o manual para mais opções).
Note que para quebrar a linha, e ficar mais legível, basta colcoar uma \ logo antes da quebra que o programa interpreta como estivesse tudo na mesma linha
singularity run /opt/images/NP3/ms_workflow/np3_ms_workflow.sif run \
-n NPTrial \
-o /home/marie.curie/tmps/ \
-m /home/marie.curie/Documentos/NPTest/marine_bacteria_lib_metadata.csv \
-d /home/luiz.alves/Documentos/NPTest/mzxml \
-t 1,2 \
-v 10
Arquivo final
Ao final, juntando tudo, temos arquivo como o abaixo que pode ser colocando em um arquivo my_np3_awsome_run.sh e ser enviado ao slurm com o comando
sbatch my_np3_awsome_run.sh
Conteúdo final do my_np3_awsome_run.sh:
#!/bin/bash
#SBATCH --job-name=NP3_MS_WORKFLOW_your_intuitive_name
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --partition=short-cpu
#SBATCH --mem-per-cpu=4G
# Execute a imagem Singularity com o comado NP3
singularity run /opt/images/NP3/ms_workflow/np3_ms_workflow.sif run \
-n NPTrial \
-o /home/marie.curie/tmps/ \
-m /home/marie.curie/Documentos/NPTest/marine_bacteria_lib_metadata.csv \
-d /home/luiz.alves/Documentos/NPTest/mzxml \
-t 1,2 \
-v 10
Usando o Job Composer do Onpen OnDemand (OOD)
O Open OnDemand é uma plataforma permite aos usuários executar aplicativos e jobs em sistemas de cluster remotos. Com o Job Composer, um componente integrado da plataforma Open OnDemand, os usuários podem facilmente criar e enviar jobs para o sistema de gerenciamento de fila. Neste tutorial passo a passo, você terá um exemplo de como usar o Job Composer para criar jobs para executar o NP³ MS_WORKFLOW.
-
Na interface inicial do OOD, após o login, entre na seção Job Composer dentro da aba jobs para acessar a ferramenta.

-
Clique no botão “New Job” na parte superior esquerda da tela para criar uma nova tarefa. Você pode escolher um template padrão ou um pré-criado. Para este exemplo, vamos no “From Default Template”.
-
A direita da tela, você verá os quadros “Job Details” e “Submit Script” com os detalhes do arquivo padrão. Para modificá-lo e preenchê-lo, clique em “Open Editor” e abra o editor de texto (provavelmente ele virá em uma nova aba).
-
Aqui você terá um editor de texto onde poderá escrever os detalhes da sua tarefa. Realize as modificações necessárias e clique em Save no canto superior esquerdo, em seguida feche a aba.
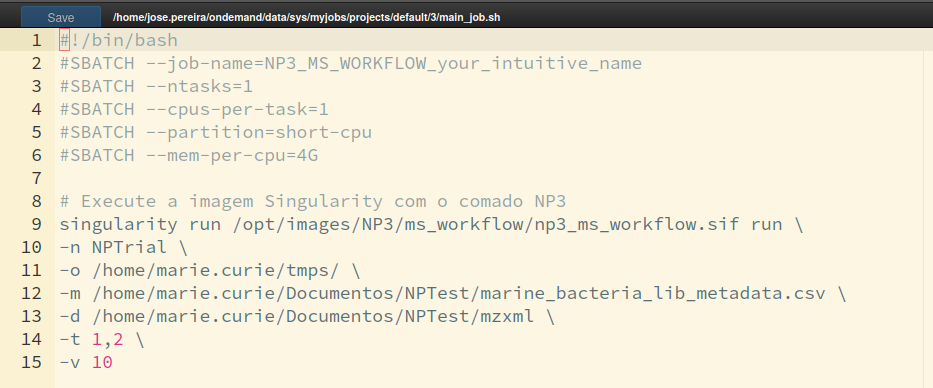
-
Se você for realizar tarefas semelhantes no futuro, é uma boa ideia criar um template. Para fazer isso, clique no botão “Create Template”.
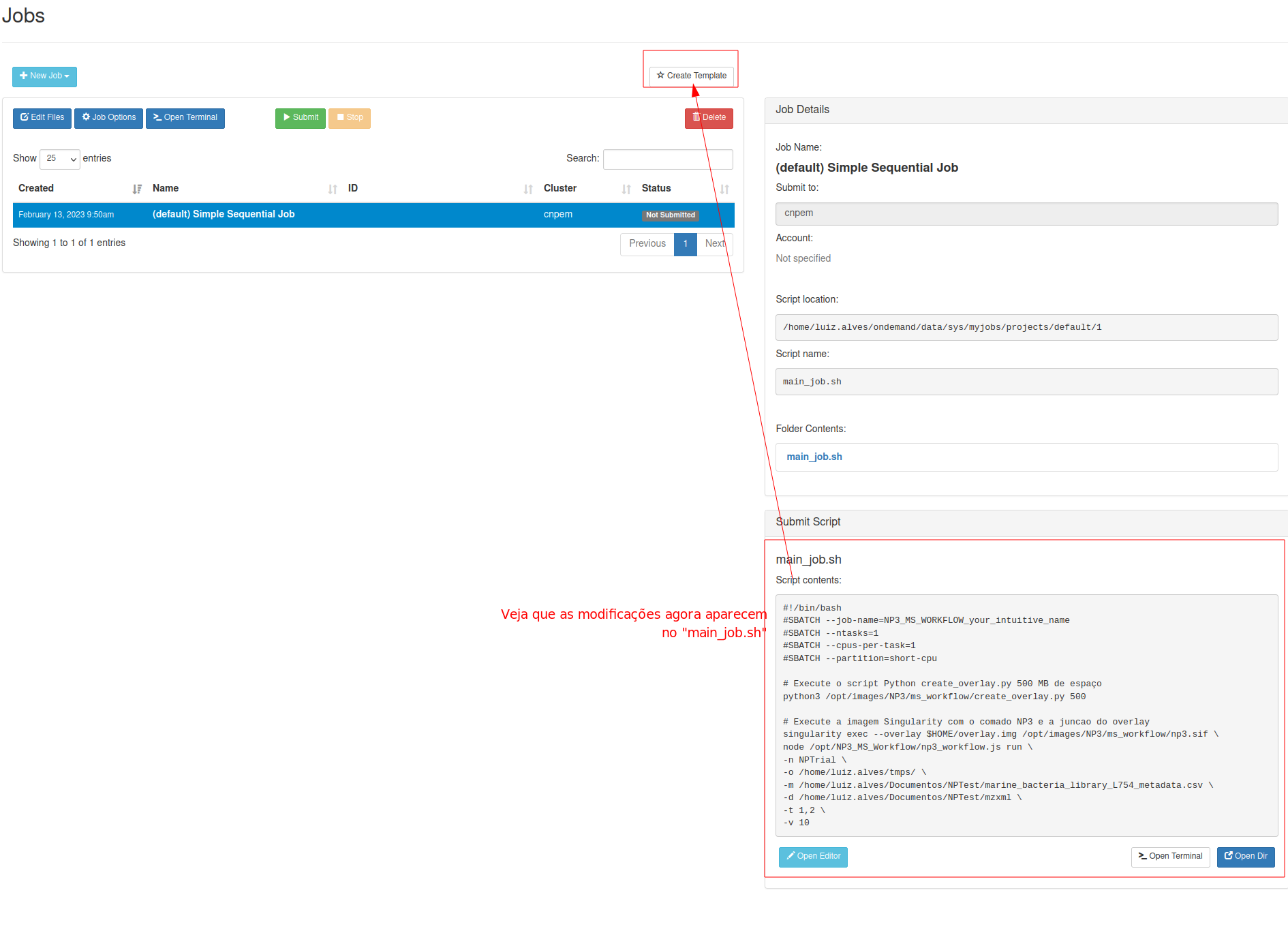
-
5.1. Preencha ou modifique os dados do fomulário e clique em Save
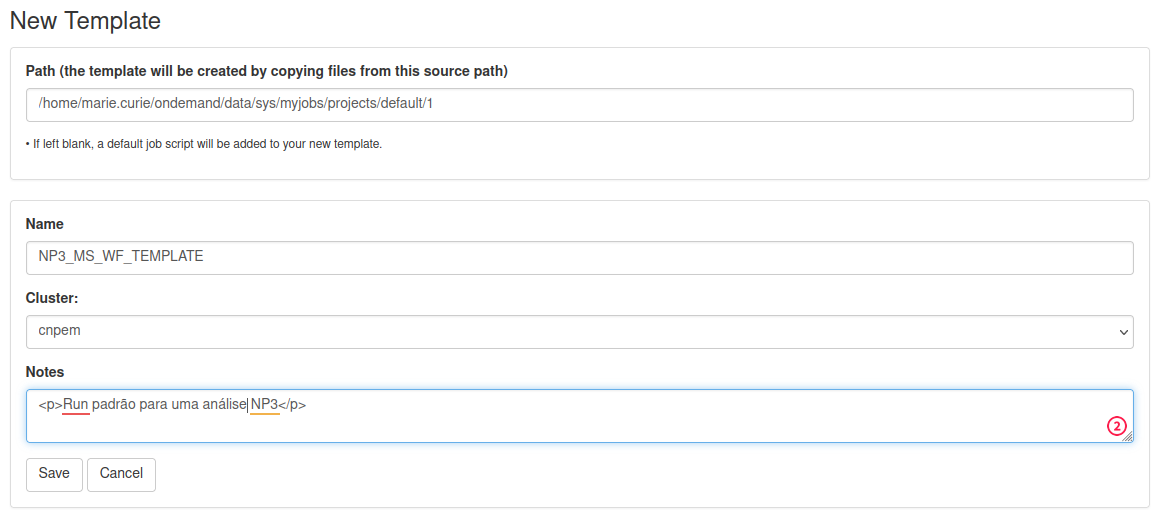
-
5.2 Agora você poderá criar tarefas a partir deste template padrão, realizando apenas as modificações necessárias como visto nos itens 3 e 4.
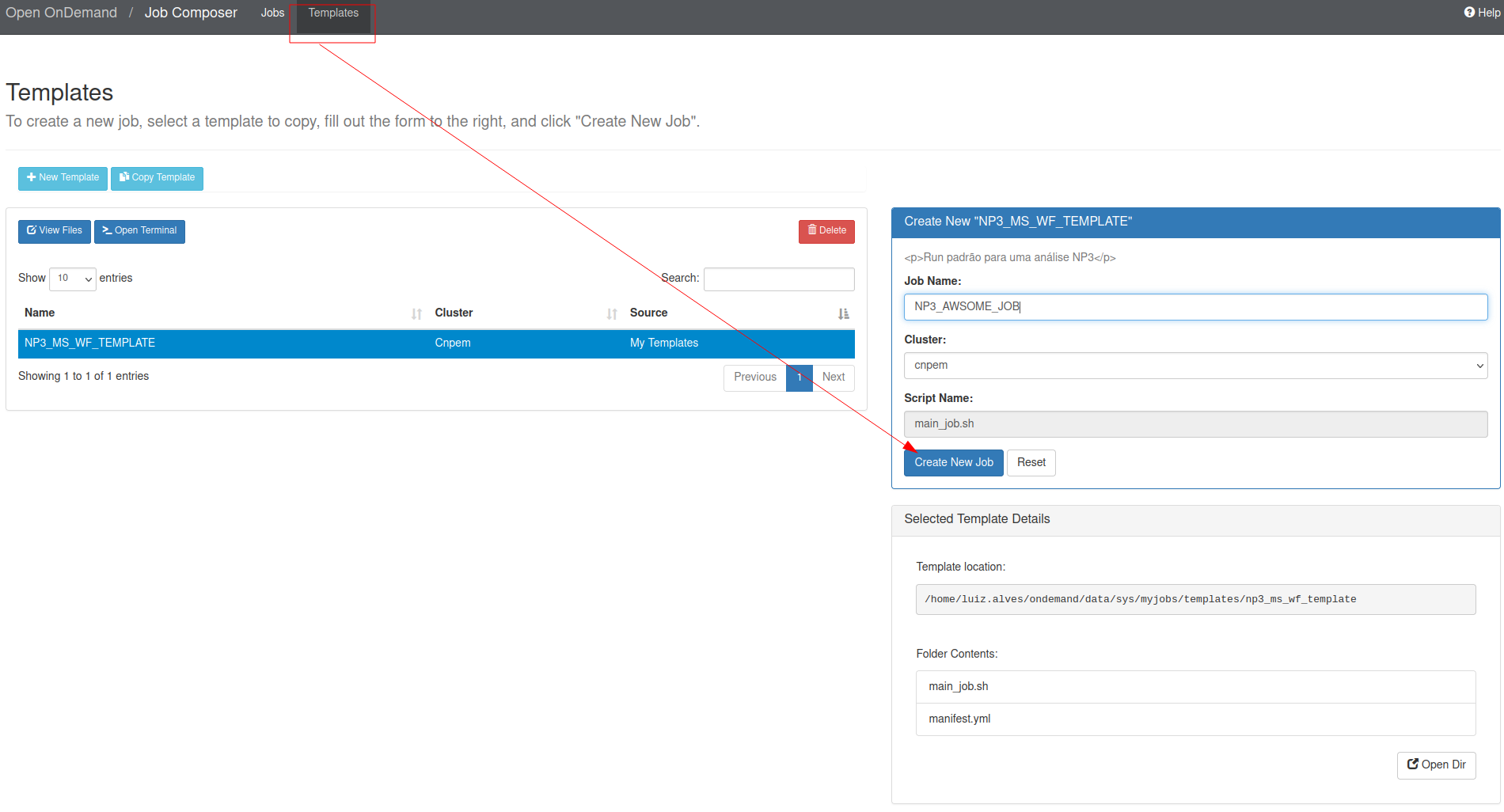
-
-
Voltando ao Job Composer, agora é escolher e conferir o job criado (fazendo mais alterações se necessário) e clicar no botão verde >Submit.
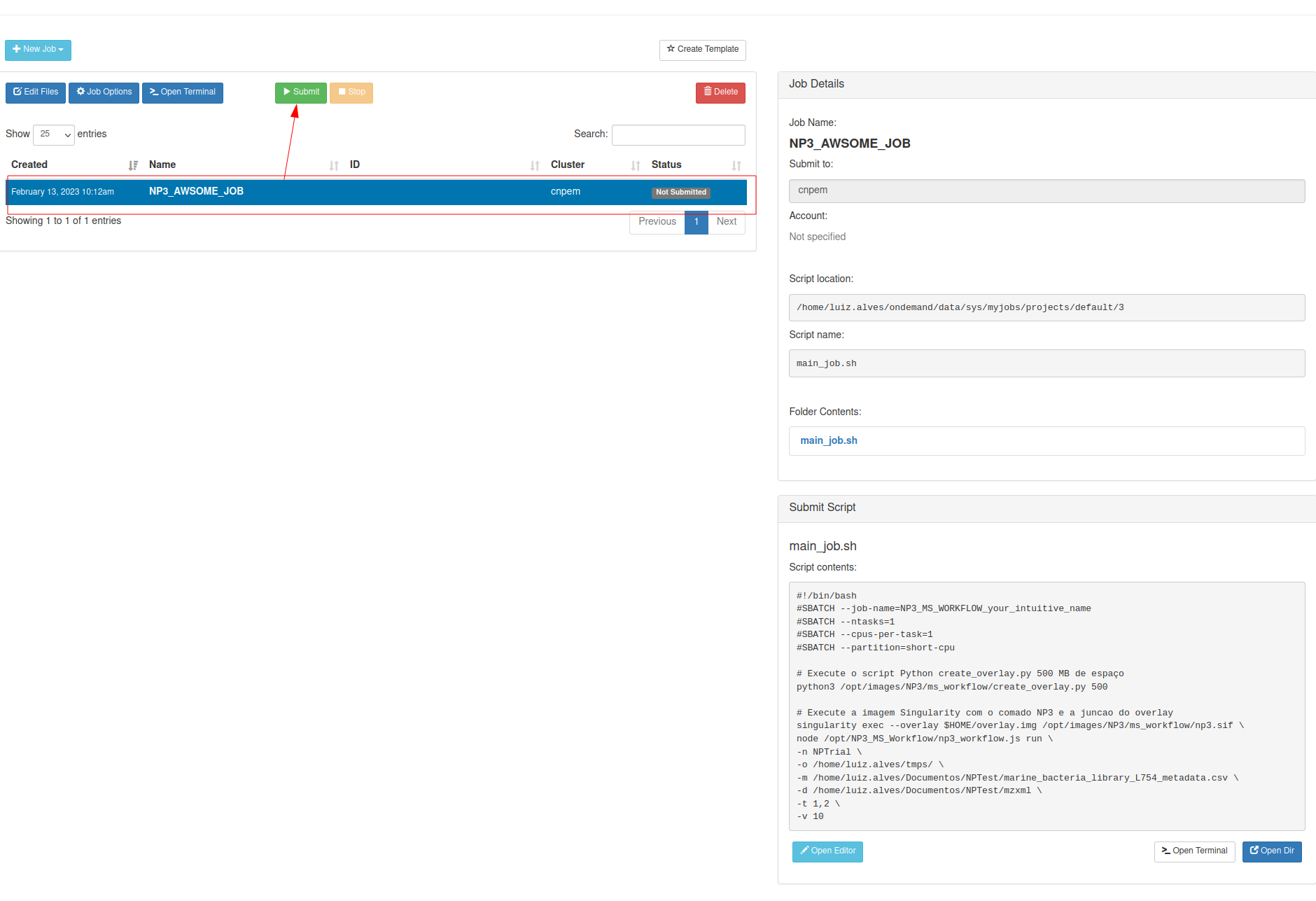
-
Você pode acompanhar o status do seu job na seção “Active Jobs” na aba “Jobs”. Aqui, você pode ver uma lista de todos os jobs ativos e já concluídos.
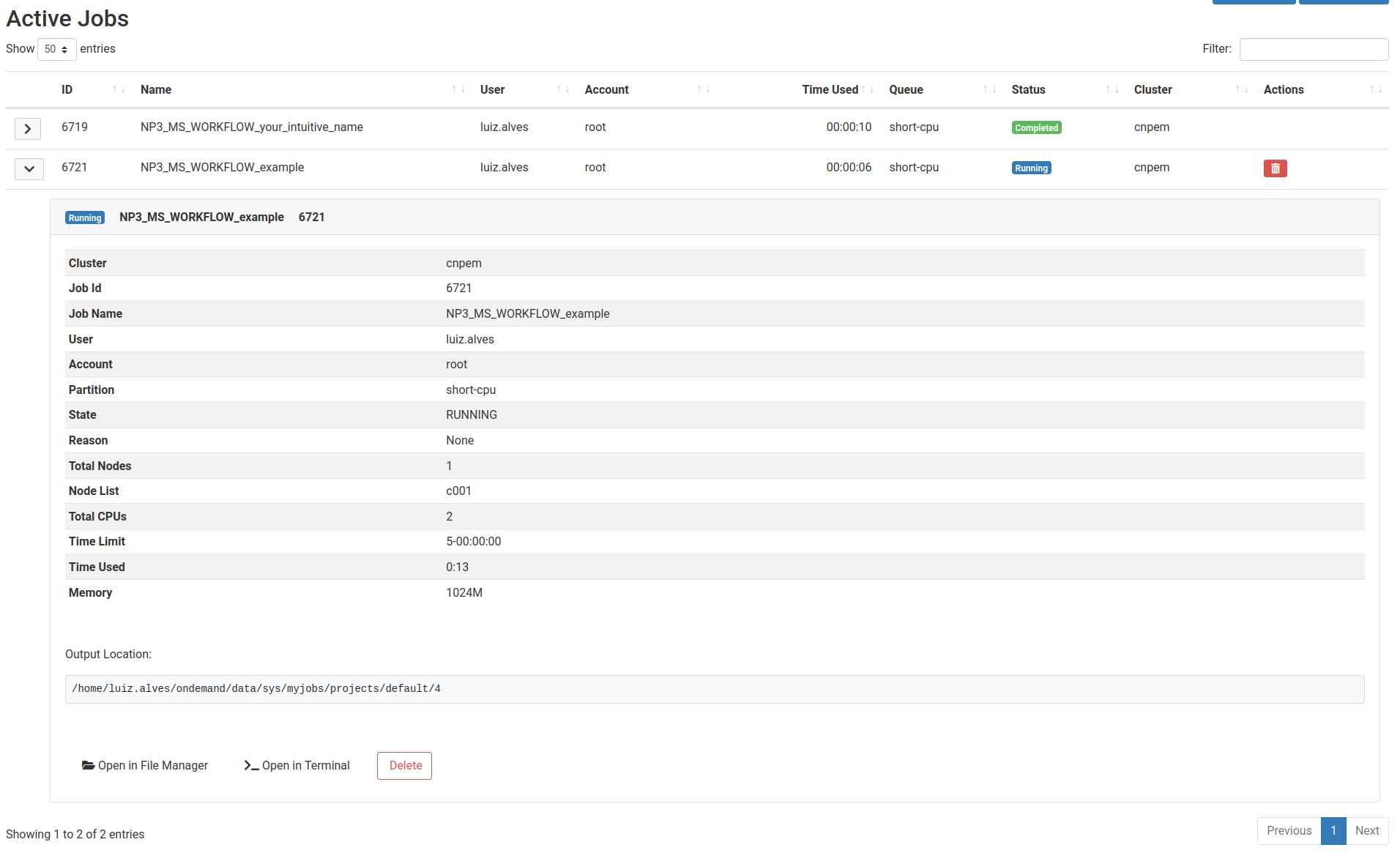
-
Para ver a saída do terminal durante o processamento do job, clique no botão Open in File Manager. Esta saída estará escrita no arquivo “slurm-xxxx.out”.
-
E os arquivos gerados pelo processamento do NP³? Eles estarão na pasta indicada pelo parâmetro “-o” ou “–output_path” no RUN submetido.
OMERO CLI
O OMERO-CLI é um conjunto de ferramentas de linha de comando baseado em Python, feito para interagir com servidores OMERO. Ele permite que os usuários realizem várias operações, como autenticação, upload/download, import/export, gerenciamento de dados e muito mais, diretamente do terminal.
Para mais informações, acesse a documentação oficial do OMERO-CLI em https://omero.readthedocs.io/en/stable/users/cli/overview.html.
Carregando o módulo
Para habilitar o OMERO-CLI no HPCC Marvin, você deve carregar o módulo omero:
module load omero
As versões disponíveis do OMERO-CLI no HPCC Marvin são:
omero/5.22 (D)
(D) indica a versão padrão.Para acessar a documentação do módulo, use:
module help omero
Para acessar a documentação completa dos parâmetros do OMERO Downloader, execute:
omero --help
Realizando login
O OMERO-CLI requer autenticação para acessar os dados armazenados no repositório. Para realizar o login, use o comando omero login:
omero login
Dados como servidor, usuário e senha serão solicitados interativamente. Você também pode passar todos eles como argumentos para evitar a solicitação interativa:
omero login -s omero-lnbio.cnpem.br -u <user_name> -w <senha>
Autenticando para o OMERO Downloader
O OMERO Downloader requer autenticação para acessar os dados armazenados no repositório. No entanto, ao informar usuário e senha diretamente no comando, essas informações podem ficar visíveis na listagem de processos do sistema. Para evitar esse cenário, recomenda-se realizar a autenticação previamente com o comando omero login e, em seguida, executar o OMERO Downloader utilizando uma chave de sessão por meio da opção -k.
Após o login, uma chave de sessão será gerada e armazenada localmente. Para usar essa chave de sessão com o OMERO Downloader, você pode verificar as suas chaves de sessão usando:
omero sessions list
Para mais detalhes, consulte a página de documentação do OMERO Downloader.
OMERO.downloader
O repositório de imagens OMERO é uma plataforma para armazenamento, gerenciamento e compartilhamento de imagens científicas, especialmente de microscopia.
O OMERO.downloader é uma ferramenta de linha de comando que serve para baixar em massa imagens e metadados de um repositório OMERO, preservando a estrutura de pastas dos datasets e projetos. Essa ferramenta é útil para obter cópias locais das imagens armazenadas no repositório.
Para mais informações sobre o OMERO.downloader, acesse https://github.com/ome/omero-downloader.
Carregando o modulo
Para habilitar o OMERO.downloader no HPCC Marvin, você deve carregar o módulo omero-downloader:
module load omero-downloader
As versões disponíveis do OMERO.downloader no HPCC Marvin são:
omero-downloader/0.2.2 (D)
(D) indica a versão padrão.Para acessar a documentação do módulo, use:
module help omero-downloader
Para acessar a documentação completa dos parâmetros do OMERO.downloader, execute:
omero-downloader --help
Realizando login
O OMERO.downloader requer autenticação para acessar os dados armazenados no repositório. No entanto, ao informar usuário e senha diretamente no comando, essas informações podem ficar visíveis na listagem de processos do sistema. Para evitar esse cenário, recomenda-se realizar a autenticação previamente com o comando omero login e, em seguida, executar o OMERO.downloader utilizando uma chave de sessão por meio da opção -k.
Para isso, carregue o módulo do OMERO CLI:
ml load omero
Em seguida, execute o comando de login:
omero login -s omero-lnbio.cnpem.br -u <user_name>
Você será solicitado a inserir senha. Após o login, uma chave de sessão será gerada e armazenada localmente. Para usar essa chave de sessão com o OMERO.downloader.
Para verificar as suas chaves de sessão, use:
omero sessions list
Você verá algo como isso:
$ omero sessions list
Server | User | Group | Session | Active | Started
----------------------+----------+-------+--------------------------------------+-----------+--------------------------
omero-lnbio.cnpem.br | analista | LIB | b22d1f3c-e7f3-4cb4-afea-4ce656d82dab | Logged in | Mon Mar 2 15:29:09 2026
(1 row)
No exemplo acima, você utilizaria a chave de sessão b22d1f3c-e7f3-4cb4-afea-4ce656d82dab com a opção -k do OMERO.downloader.
Baixando dados
O comando base para baixar dados com o OMERO.downloader é o seguinte:
omero-downloader -b <output_dir> -k <session_key> -f <file> <type>:<ID>
Substitua as informações indicadas entre colchetes angulares (
<...>):
<output_dir>: diretório onde os dados serão baixados. É importante que a pasta já exista.<user_name>: seu nome de usuário institucional, no formatonome.sobrenome.<password>: sua senha institucional.<file>: formato do arquivo a ser baixado. O formato recomendado éome-tiff; outros formatos estão disponíveis na documentação de parâmetros do OMERO.downloader.<type>: tipo de objeto que você deseja baixar:Image: imagem individualPlate: placa de experimentos multi-poçosScreen: conjunto de placasDataset: conjunto de arquivos de imagemProject: conjunto de datasets
<ID>:identificador numérico do objeto no OMERO..
Para baixar uma imagem, use:
omero-downloader -b /home/marie.curie/pasta_destino -k <session_key> -f ome-tiff Image:123
Para baixar um Dataset, use:
omero-downloader -b /home/marie.curie/pasta_destino -k <session_key> -f ome-tiff Dataset:123
Para baixar um projeto, use:
omero-downloader -b /home/marie.curie/pasta_destino -k <session_key> -f ome-tiff Project:123
Para descobrir o número de identificação (ID) de uma imagem, dataset ou projeto, acesse a plataforma https://omero-lnbio.cnpem.br. Nela, é possível visualizar a organização dos projetos, datasets e imagens aos quais você tem acesso. Ao selecionar o item desejado, o ID correspondente será exibido no painel à direita, na guia “General”.
Pairix
O Pairix é uma ferramenta para indexação e consulta eficiente de arquivos .pairs, usados em análises de dados Hi-C e genômica estrutural.
Baseado no Tabix, o Pairix permite buscas rápidas por intervalos genômicos em arquivos de pares, suportando consultas por uma ou duas regiões genômicas.
É amplamente utilizado em pipelines de análise e visualização de dados de interação cromossômica.
Para mais informações, acesse: https://github.com/4dn-dcic/pairix
Carregando o módulo
Para habilitar o pairix no HPCC Marvin, você deve carregar o módulo pairix:
module load pairix
As versões disponíveis do pairix no HPCC Marvin são:
pairix/0.3.9 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help pairix
Executando o módulo
Além do executável pairix, o módulo contém outros binários auxiliares como bgzip, pairs_merger e streamer_1d. Execute-os para ter mais detalhes sobre o uso e parâmetros necessários e opcionais.
Exemplos de utilização do pairix para indexar:
pairix textfile.gz # for recognized file extension
pairix -p <preset> textfile.gz
pairix -s<chr1_column> [-d<chr2_column>] -b<pos1_start_column> -e<pos1_end_column> [-u<pos2_start_column> -v<pos2_end_column>] [-T] textfile.gz # u, v is required for full 2d query.
Exemplo para realizar query:
pairix textfile.gz region1 [region2 [...]] ## region is in the following format.
# for 1D indexed file
pairix textfile.gz '<chr>:<start>-<end>' '<chr>:<start>-<end>' ...
# for 2D indexed file
pairix [-a] textfile.gz '<chr1>:<start1>-<end1>|<chr2>:<start2>-<end2>' ... # make sure to quote so '|' is not interpreted as a pipe.
pairix [-a] textfile.gz '*|<chr2>:<start2>-<end2>' # wild card is accepted for 1D query on 2D indexed file
pairix [-a] textfile.gz '<chr1>:<start1>-<end1>|*' # wild card is accepted for 1D query on 2D indexed file
Consulte sobre a utilização, opções e parâmetros disponíveis com pairix -h e informações mais detalhadas na no repositório do pairix.
Submetendo jobs
A execução do pairix no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo pairix.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=pairix
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=2
#SBATCH --mem-per-cpu=2GB
module load pairix/0.3.9
INPUT_PAIRS="pairs_file.gz"
REGION_1="<chr>:<start>-<end>"
REGION_2="<chr>:<start>-<end>"
pairix "$INPUT_PAIRS" "$REGION_1" "$REGION_2"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch pairix.sh
Para mais detalhes sobre os parâmetros do pairix, use:
pairix -h
pairtools
O pairtools é um conjunto de ferramentas em linha de comando e biblioteca Python desenvolvido para processar e manipular dados de pares de leituras provenientes de experimentos Hi-C. Ele realiza etapas essenciais do pipeline, como emparelhamento de leituras mapeadas, filtragem, deduplicação e indexação, gerando arquivos .pairs padronizados que descrevem interações cromossômicas individuais. O pairtools é altamente eficiente e modular, podendo ser integrado a pipelines maiores para análise de dados genômicos em larga escala.
Para mais informações e documentação completa, acesse: https://pairtools.readthedocs.io/en/latest/index.html
Carregando o módulo
Para habilitar o pairtools no HPCC Marvin, você deve carregar o módulo pairtools:
module load pairtools
As versões disponíveis do pairtools no HPCC Marvin são:
pairtools/1.1.3 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help pairtools
Executando o módulo
Modelo de uso do pairtools via linha de comando:
pairtools [OPTIONS] COMMAND [ARGS]...
Ao executar pairtools -h, é possível consultar a lista de comandos disponíveis:
Commands:
dedup Find and remove PCR/optical duplicates.
filterbycov Remove pairs from regions of high coverage.
flip Flip pairs to get an upper-triangular matrix.
header Manipulate the .pairs/.pairsam header
markasdup Tag all pairs in the input file as duplicates.
merge Merge .pairs/.pairsam files.
parse Find ligation pairs in .sam data, make .pairs.
parse2 Extracts pairs from .sam/.bam data with complex walks,...
phase Phase pairs mapped to a diploid genome.
restrict Assign restriction fragments to pairs.
sample Select a random subset of pairs in a pairs file.
scaling Calculate pairs scalings.
select Select pairs according to some condition.
sort Sort a .pairs/.pairsam file.
split Split a .pairsam file into .pairs and .sam.
stats Calculate pairs statistics.
Consulte todos informações completas sobre os comandos na página de referência de CLI na documentação oficial do pairtools.
Submetendo jobs
A execução do pairtools no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo pairtools.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=pairtools
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=4GB
module load pairtools/1.1.3
PAIRS_PATH="/caminho/para/arquivo.pairs"
OUTPUT="/caminho/para/output.tsv"
pairtools dedup -o "$OUTPUT" "$PAIRS_PATH"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch pairtools.sh
Para mais detalhes sobre os parâmetros de cada comando do pairtools, use:
pairtools [COMMAND] -h
Phenix
O Phenix é um conjunto abrangente de ferramentas para a determinação e análise de estruturas macromoleculares, amplamente utilizado na comunidade de biologia estrutural. Ele oferece uma variedade de programas para refinamento, validação e modelagem de estruturas de proteínas e ácidos nucleicos, facilitando a interpretação de dados experimentais obtidos por cristalografia de raios X, microscopia eletrônica e outras técnicas.
Para mais informações sobre o Phenix, acesse https://phenix-online.org/documentation/.
Carregando o módulo
O Phenix está disponível dentro do módulo scipion/3.8.3. Para utilizá-lo no HPCC Marvin, você deve carregar o módulo scipion/3.8.3:
module load scipion/3.8.3
Para acessar a documentação do modulo, utilize:
module help scipion/3.8.3
Como executar o Phenix no Open OnDemand
Para executar o Phenix, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o Phenix:
# Habilitar o módulo
module load scipion/3.8.3
# Iniciar o Phenix com interface gráfica
phenix
Para mais detalhes sobre os parâmetros do Phenix, use:
phenix --help
Procheck
O Procheck é um pacote de software para avaliar a qualidade geométrica de estruturas de proteínas.
Ele é comumente utilizado para verificar parâmetros como:
- Geometria dos resíduos (ângulos phi/psi)
- Validação de ligações de hidrogênio e distâncias
- Conformidade de resíduos com estruturas cristalográficas conhecidas
- Geração de gráficos de qualidade e relatórios detalhados
Para mais informações sobre o PROCHECK, acesse https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/
Carregando o módulo
Para habilitar o PROCHECK no HPCC Marvin, você deve carregar o módulo procheck:
module load procheck
As versões disponíveis do Procheck no HPCC Marvin são:
procheck/3.5.4 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help procheck
Configurando o módulo
O PROCHECK inclui várias ferramentas e scripts, como:
- procheck — avaliação geral de estruturas
- procheck-nmr — avaliação de estruturas NMR
- procheck-comp — comparação de conformações
Ao carregar o módulo com module load procheck, é automaticamente executado um script para configuração de variáveis e alias que chamarão os diferentes binários do pacote. O script verifica qual o shell do usuário (bash/zsh ou csh/tcsh) e realiza a configuração de acordo.
Os alias atualmente configurados pelo script são:
prodir='/opt/images/apps/procheck/v3.5.4'
export prodir
alias procheck=$prodir'/procheck.scr'
alias procheck_comp=$prodir'/procheck_comp.scr'
alias procheck_nmr=$prodir'/procheck_nmr.scr'
alias proplot=$prodir'/proplot.scr'
alias proplot_comp=$prodir'/proplot_comp.scr'
alias proplot_nmr=$prodir'/proplot_nmr.scr'
alias aquapro=$prodir'/aquapro.scr'
alias gfac2pdb=$prodir'/gfac2pdb.scr'
alias viol2pdb=$prodir'/viol2pdb.scr'
alias wirplot=$prodir'/wirplot.scr'
Para mais informações sobre os diferentes scripts e programas do Procheck, consulte o Manual e o NMR Manual
Submetendo jobs
A execução do Procheck no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo procheck.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=procheck
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=2GB
module load procheck/3.5.4
EXAMPLE_FILE="/data/pdb/p1amt.pdb" # the coordinates file in Brookhaven format
CHAIN="A" # an optional one-letter chain-ID
RESOLUTION=1.5 # a real number giving the resolution of the structure
procheck "$EXAMPLE_FILE" "$CHAIN" "$RESOLUTION"
O script acima funciona para o procheck básico. Caso queira executar o procheck_nmr ou procheck_comp, por exemplo, adapte as variáveis e argumentos de acordo com seus respectivos usos.
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch procheck.sh
Para mais detalhes sobre os parâmetros do Procheck, use:
procheck --help
pyGenomeTracks
O pyGenomeTracks é uma ferramenta para gerar visualizações integradas de dados genômicos a partir de múltiplas faixas (tracks), como Hi-C, ChIP-seq, RNA-seq, entre outros.
Ele permite criar gráficos de alta qualidade combinando várias camadas de informação em uma única figura, facilitando a interpretação e comparação de diferentes experimentos genômicos.
Para mais informações, acesse: https://github.com/deeptools/pyGenomeTracks
Carregando o módulo
Para habilitar o pyGenomeTracks no HPCC Marvin, você deve carregar o módulo pyGenomeTracks:
module load pyGenomeTracks
As versões disponíveis do pyGenomeTracks no HPCC Marvin são:
pyGenomeTracks/3.9 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help pyGenomeTracks
Executando o módulo
Para executar o pyGenomeTracks, é necessário um arquivo de configuração que descreva as tracks (faixas de dados).
A maneira mais fácil de criar esse arquivo é usando o programa make_tracks_file, que gera uma configuração padrão facilmente editável.
Comando para criar o arquivo de configuração:
make_tracks_file --trackFiles <file1.bed> <file2.bw> ... -o tracks.ini
O make_tracks_file identifica automaticamente o tipo de arquivo com base em sua extensão.
Comando para gerar o gráfico de uma região genômica:
pyGenomeTracks --tracks tracks.ini --region chr2:10,000,000-11,000,000 --outFileName nice_image.pdf
A opção --outFileName define o formato da imagem de saída. Se for usado .pdf, o resultado será um arquivo PDF. Os formatos disponíveis são pdf, png e svg.
Submetendo jobs
A execução do pyGenomeTracks no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo pyGenomeTracks.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=pyGenomeTracks
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem-per-cpu=2GB
module load pyGenomeTracks/3.9
TRACKS="/caminho/para/tracks/file"
REGION="chr2:10,000,000-11,000,000"
OUTPUT_FILE="out.pdf"
pyGenomeTracks --tracks "$TRACKS" --region "$REGION" --outFileName "$OUTPUT_FILE"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch pyGenomeTracks.sh
Para mais detalhes sobre os parâmetros do pyGenomeTracks, use:
pyGenomeTracks -h
Relion
O Relion (REgularised LIkelihood OptimisatioN) é um software amplamente utilizado para o processamento de dados de microscopia eletrônica de partículas únicas (cryo-EM). Ele implementa métodos estatísticos avançados para a reconstrução tridimensional de estruturas moleculares a partir de imagens bidimensionais, permitindo a obtenção de mapas de alta resolução. O Relion é conhecido por sua capacidade de lidar com grandes conjuntos de dados e por sua abordagem robusta para a classificação e refinamento de partículas.
Para mais informações sobre o Relion, acesse https://relion.readthedocs.io/en/release-5.0/.
Carregando o módulo
O RELION está disponível dentro do módulo scipion/3.8.3. Para utilizá-lo no HPCC Marvin, você deve carregar o módulo scipion/3.8.3:
module load scipion/3.8.3
Para acessar a documentação do modulo, utilize:
module help scipion/3.8.3
Como executar o Relion no Open OnDemand
Para executar o Relion, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o Relion:
# Habilitar o módulo
module load scipion/3.8.3
# Iniciar o Relion com interface gráfica
relion
Para mais detalhes sobre os parâmetros do Relion, use:
relion --help
Scipion
O Scipion é uma plataforma de software integrada para processamento de dados de microscopia eletrônica de partículas únicas e tomografia (Cryo-EM). Ele fornece uma interface unificada para vários pacotes de software, facilitando o fluxo de trabalho desde a aquisição de dados até a reconstrução 3D e análise estrutural.
Para mais informações sobre o Scipion, acesse https://scipion-em.github.io/docs/release-3.0.0/index.html.
Carregando o módulo
Carregando o módulo
Para habilitar o Scipion no HPCC Marvin, você deve carregar o módulo scipion:
module load scipion
As versões disponíveis do Scipion no HPCC Marvin são:
scipion/3.8.3 (D)scipion/3.0.12
(D) indica a versão padrão.
A versão scipion/3.8.3 inclui outros programas integrados, como:
3dmod,
cisTEM,
Etomo,
IsoNet,
Phenix e
Relion.
Para acessar a documentação do modulo, utilize:
module help scipion
Como executar o Scipion no Open OnDemand
Para executar o Scipion, são necessários os seguintes passos:
-
Acesse o Open OnDemand do HPCC Marvin em https://marvin.cnpem.br/.
-
Em
Interactive Apps, abra umaVNC. -
No formulário da VNC, selecione uma das partições
gui-gpu-smallougui-gpu-bige defina o número de horas, número de GPUs e número de CPUs conforme necessário. Clique emLaunch. -
Uma nova janela será aberta com a VNC. Aguarde até que a VNC esteja ativa (
Running) e clique emLaunch VNC. -
Uma vez que a VNC estiver ativa, abra um terminal dentro da VNC.
-
No terminal, execute o seguinte comando para iniciar o Scipion:
# Habilitar o módulo
module load scipion
# Iniciar o Scipion com interface gráfica
scipion
Para mais detalhes sobre os parâmetros do Scipion, use:
scipion --help
seqtk
O seqtk é um conjunto de ferramentas leves em linha de comando para o processamento rápido de arquivos FASTA e FASTQ. Desenvolvido em C, ele é altamente eficiente e indicado para tarefas comuns de manipulação de sequências, como conversão entre formatos, amostragem aleatória, filtragem, recorte, cálculo de estatísticas e reversão de sequências. O seqtk é amplamente utilizado em pipelines de bioinformática por sua velocidade, simplicidade e fácil integração com outros utilitários de linha de comando.
Para mais informações e documentação completa, acesse: https://github.com/lh3/seqtk/tree/master
Carregando o módulo
Para habilitar o seqtk no HPCC Marvin, você deve carregar o módulo seqtk:
module load seqtk
As versões disponíveis do seqtk no HPCC Marvin são:
seqtk/1.5 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help seqtk
Executando o módulo
Modelo de uso do seqtk via linha de comando:
seqtk [OPTIONS] COMMAND [ARGS]...
Ao executar seqtk -h, é possível consultar a lista de comandos disponíveis:
Command:
seq common transformation of FASTA/Q
size report the number sequences and bases
comp get the nucleotide composition of FASTA/Q
sample subsample sequences
subseq extract subsequences from FASTA/Q
fqchk fastq QC (base/quality summary)
mergepe interleave two PE FASTA/Q files
split split one file into multiple smaller files
trimfq trim FASTQ using the Phred algorithm
hety regional heterozygosity
gc identify high- or low-GC regions
mutfa point mutate FASTA at specified positions
mergefa merge two FASTA/Q files
famask apply a X-coded FASTA to a source FASTA
dropse drop unpaired from interleaved PE FASTA/Q
rename rename sequence names
randbase choose a random base from hets
cutN cut sequence at long N
gap get the gap locations
listhet extract the position of each het
hpc homopolyer-compressed sequence
telo identify telomere repeats in asm or long reads
Para mais detalhes sobre os parâmetros de cada comando do seqtk, use:
seqtk [COMMAND] -h
Também consulte os exemplos de uso no repositório do seqtk.
Submetendo jobs
A execução do seqtk no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo seqtk.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=seqtk
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --mem-per-cpu=2GB
module load seqtk/1.5
INPUT_FASTQ="/caminho/para/input.fq.gz"
OUTPUT_FASTA="/caminho/para/output.fa"
# Convert FASTQ to FASTA
seqtk seq -a "$INPUT_FASTQ" > "$OUTPUT_FASTA"
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch seqtk.sh
TCRmodel2
O TCRmodel2 é um programa de predição de estrutura de proteínas baseado em adaptações do AlphaFold2. Apresenta alta acurácia de predição do complexo TCR:pMHC, assim como, estruturas unbound de TCRs. O tempo de predição (~15 minutos por complexo para as regiões variáveis) também é reduzido em comparação a outros modelos. As predições já possuem resíduos renumerados para o esquema AHo e cadeias renomeadas para MHC (A), peptídeo (C), TCRalpha (D) e TCRbeta (E).
Para mais informações sobre TCRmodel2, acesse https://github.com/piercelab/tcrmodel2.
Carregando o módulo
Para habilitar o TCRmodel2 no HPCC Marvin, você deve carregar o módulo tcrmodel2:
module load tcrmodel2
Para acessar a documentação do modulo, utilize:
module help tcrmodel2
Executando o módulo
O TCRmodel2 oferece dois principais scripts:
run_tcrmodel2.py: predição do complexo TCR:pMHC que pode ser chamado comtcrmodel2run_tcrmodel2_ub_tcr.py: predição de estruturas unbound de TCRs que pode ser chamado comtcrmodel2_ub_tcr
Ambos scripts contém diferentes flags. Para obter informações sobre o uso de cada um, execute:
tcrmodel2 --help #or
tcrmodel2_ub_tcr --help
O banco de dados e pesos do AF2 estão na pasta
/public do HPC e são necessários para execução dos scripts.
Submetendo jobs
A execução do TCRmodel2 no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Para modelar apenas o TCR unbound, crie um arquivo de script, por exemplo tcrmodel2.sh, com o seguinte conteúdo:
#!/bin/bash
#SBATCH --job-name=tcrmodel2
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=6
#SBATCH --partition=short-gpu-small
#SBATCH --mem-per-cpu=8G
#SBATCH --gres=gpu:1g.5gb:5
#SBATCH --output=slurm.out
#SBATCH --error=slurm.error
ml load tcrmodel2
OUTPUT_DIR="/output"
TCRA="GQQVMQIPQYQHVQEGEDFTTYCNSSTTLSNIQWYKQRPGGHPVFLIQLVKSGEVKKQKRLTFQFGEAKKNSSLHITATQTTDVGTYFCAVSYGGSQGNLIFGKGTKLSVKP"
TCRB="DGGITQSPKYLFRKEGQNVTLSCEQNLNHDAMYWYRQDPGQGLRLIYYSQIVNDFQKGDIAEGYSVSREKKESFPLTVTSAQKNPTAFYLCASSIRSTDTQYFGPGTRLTVLE"
tcrmodel2_ub_tcr --job_id=test --output_dir=$OUTPUT_DIR --tcra_seq=$TCRA --tcrb_seq=$TCRB --ori_db=/database/ --tp_db=/opt/tcrmodel2/data/databases --relax_structures=True --max_template_date=2100-01-01
Ao carregar o módulo tcrmodel2, ficarão disponíveis dois comandos (tcrmodel2 e tcrmodel2_ub_tcr) para modelar, respectivamente, o complexo inteiro ou somente TCRab. Lembrando que, para modelar o complexo inteiro, siga as instruções do repositório do TCRmodel2 para configurar as flags e substitua o comando tcrmodel2_ub_tcr por tcrmodel2 no script do slurm.
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch tcrmodel2.sh
Trimmomatic
O Trimmomatic é uma ferramenta em linha de comando amplamente utilizada para o pré-processamento de dados de sequenciamento de DNA provenientes de plataformas Illumina. Ela realiza etapas fundamentais de limpeza das leituras (reads), incluindo remoção de adaptadores, filtragem por qualidade, corte de regiões de baixa qualidade e eliminação de leituras curtas, garantindo que apenas dados de alta confiabilidade sejam utilizados em análises posteriores. Altamente flexível e eficiente, suporta tanto dados single-end quanto paired-end, além de permitir o ajuste fino de parâmetros conforme o protocolo experimental.
Para mais informações, acesse: https://github.com/usadellab/Trimmomatic.
Carregando o módulo
Para habilitar o trimmomatic no HPCC Marvin, você deve carregar o módulo trimmomatic:
module load trimmomatic
As versões disponíveis do trimmomatic no HPCC Marvin são:
trimmomatic/0.4 (D)
(D) indica a versão padrão.Para acessar a documentação do modulo, utilize:
module help trimmomatic
Executando o módulo
Há dois modos de executar o trimmomatic, SE (Singe End) para leituras únicas, e PE (Paired End) para leituras emparelhadas.
Single end
Como executar o módulo no modo SE:
trimmomatic SE [-threads <threads>] [-phred33 | -phred64] [-trimlog <logFile>] <input> <output> <step 1> ...
Exemplo:
trimmomatic SE -threads 4 input.fastq.gz output_trimmed.fastq.gz \
ILLUMINACLIP:TruSeq3-SE.fa:2:30:10 SLIDINGWINDOW:4:20 MINLEN:36
Paired end
Como executar o módulo no modo PE:
trimmomatic PE [-threads <threads>] [-phred33 | -phred64] [-trimlog <logFile>] >] [-basein <inputBase> | <input 1> <input 2>] [-baseout <outputBase> | <unpaired output 1> <paired output 2> <unpaired output 2> <step 1> ...
Exemplo:
trimmomatic PE -threads 8 input_R1.fastq.gz input_R2.fastq.gz \
output_R1_paired.fastq.gz output_R1_unpaired.fastq.gz \
output_R2_paired.fastq.gz output_R2_unpaired.fastq.gz \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 SLIDINGWINDOW:4:20 MINLEN:36
Consulte as informações completas sobre os comandos e opções do módulo no repositório oficial do trimmomatic no GitHub e também no manual em pdf (versão 0.32).
Submetendo jobs
A execução do trimmomatic no HPCC Marvin é feita por meio de scripts de submissão no SLURM. Crie um arquivo de script, por exemplo trimmomatic.sh, com o seguinte conteúdo:
u
#!/bin/bash
#SBATCH --job-name=trimmomatic
#SBATCH --partition=short-cpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem=8G
INPUT_R1="reads_R1.fastq.gz"
INPUT_R2="reads_R2.fastq.gz"
OUTPUT_R1_PAIRED="reads_R1_paired.fastq.gz"
OUTPUT_R1_UNPAIRED="reads_R1_unpaired.fastq.gz"
OUTPUT_R2_PAIRED="reads_R2_paired.fastq.gz"
OUTPUT_R2_UNPAIRED="reads_R2_unpaired.fastq.gz"
ADAPTERS="path/to/adapters/TruSeq3-PE.fa"
module load trimmomatic/0.4
trimmomatic PE -threads ${SLURM_CPUS_PER_TASK} \
$INPUT_R1 $INPUT_R2 \
$OUTPUT_R1_PAIRED $OUTPUT_R1_UNPAIRED \
$OUTPUT_R2_PAIRED $OUTPUT_R2_UNPAIRED \
ILLUMINACLIP:${ADAPTERS}:2:30:10 SLIDINGWINDOW:4:20 MINLEN:36
Para submeter o job, salve o script e utilize o comando sbatch:
sbatch trimmomatic.sh
Sistemas
Atualmente, os seguintes sistemas estão disponíveis para os usuários do LNBio:
Para instalar ou atualizar um sistema, você deve registrar um chamado [LNBio] Suporte EDB do Jira em HPCC Marvin: Aplicativos, Programas e Sistemas.
OMERO
Esta seção apresenta o repositório de imagens OMERO do LNBio, que está disponível em https://omero-lnbio.cnpem.br.
O OMERO é uma plataforma de gerenciamento de imagens científicas desenvolvida pelo Open Microscopy Environment (OME), uma iniciativa de código aberto amplamente utilizada na comunidade científica para o armazenamento, visualização e compartilhamento de imagens de microscopia.
- Acesso pelo navegador
- Acessando os grupos
- Carregando imagens via OMERO.insight
- Anotando imagens via OMERO.web
- Baixando imagens via OMERO.downloader
Para solicitar suporte ou ajuda com o OMERO, registre um chamado na [LNBio] Suporte EDB do Jira em OMERO: Suporte ao usuário.
Acesso pelo navegador
Acesso pelo navegador 
Para acessar o OMERO pelo navegador, abra seu navegador e acesse:
https://omero-lnbio.cnpem.br
Na tela de login, use seu usuário (sem @lnbio.cnpem.br) e senha institucional.
marie.curie@lnbio.cnpem.br.
Logo, seu usuário é marie.curie.

Após o login, você verá a interface web do OMERO:
Acessando os grupos
Acessando os grupos
Após realizar o login, você poderá selecionar o grupo de trabalho no qual deseja navegar.
No canto superior esquerdo da interface, clique no ícone de 👥 (grupos) para listar todos os grupos dos quais você faz parte. Ao passar o mouse sobre um grupo, os usuários pertencentes a ele serão exibidos.
Ao clicar sobre um usuário dentro de um grupo, você poderá visualizar, anotar ou editar imagens, dependendo das permissões configuradas para aquele grupo. Por padrão, os grupos permitem apenas visualização.
Carregando imagens via OMERO.insight
O OMERO.insight é uma aplicação desktop que permite o gerenciamento e visualização de imagens armazenadas no servidor OMERO. Ele oferece uma interface gráfica amigável para navegar, visualizar e organizar suas imagens.
Instalando o OMERO.insight
Para realizar o envio das imagens à plataforma OMERO, é necessário instalar o programa OMERO.insight no computador que será utilizado para esse procedimento. O instalador está disponível em: https://www.openmicroscopy.org/omero/downloads/.

Escolha a versão mais recente disponível para o seu sistema operacional.
Configurando o OMERO.insight
- Após a instalação, abra o OMERO.insight em seu computador e clique no ícone de ferramenta (🔧), localizado no canto superior direito da janela.

- Em seguida, clique no botão (➕) para adicionar um novo endereço de plataforma OMERO;
- Na janela “Add new server”, digite o seguinte endereço
omero-lnbio.cnpem.bre clique em “⏎ OK”;
- Selecione o endereço omero-lnbio.cnpem.br e clique em “Apply” (Figura 5b);
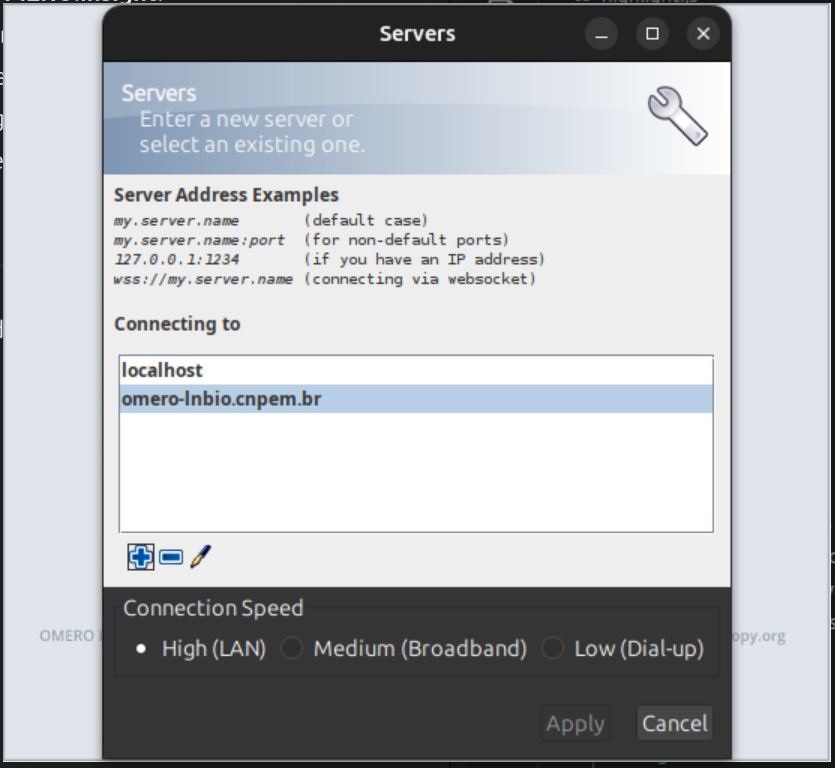
Transferindo imagens para o OMERO
Após configurar o OMERO do LNBio no OMERO.insight, utilize suas credenciais institucionais do CNPEM para fazer login.

marie.curie em “Username” e sua senha institucional em “Password”.
Após o login, você terá acesso à interface do OMERO.insight, onde poderá visualizar e gerenciar suas imagens.
Para carregar novas imagens, siga os passos abaixo:
- Clique no menu “File” e selecione “Import…”
- Na janela de importação, selecione os arquivos que deseja carregar para o OMERO, e clique em
>para colocá-los na fila de transferência.
- Uma janela para definir o projeto de destino aparecerá. Selecione o projeto desejado ou crie um novo, e clique em “Add to the Queue”.
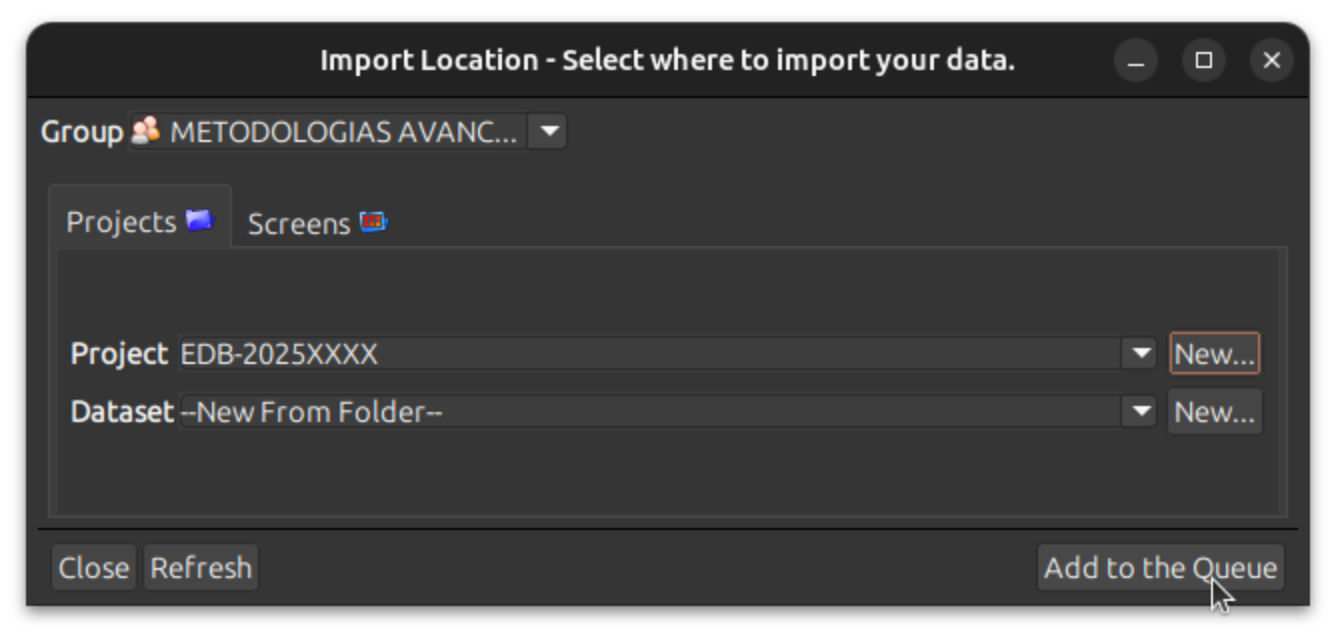
- Ajuste as configurações de importação. Desmarque opção
Override default File naming, e caso deseje acelerar o processo de transferência, ative todas as opções da áreaImport Speedup.
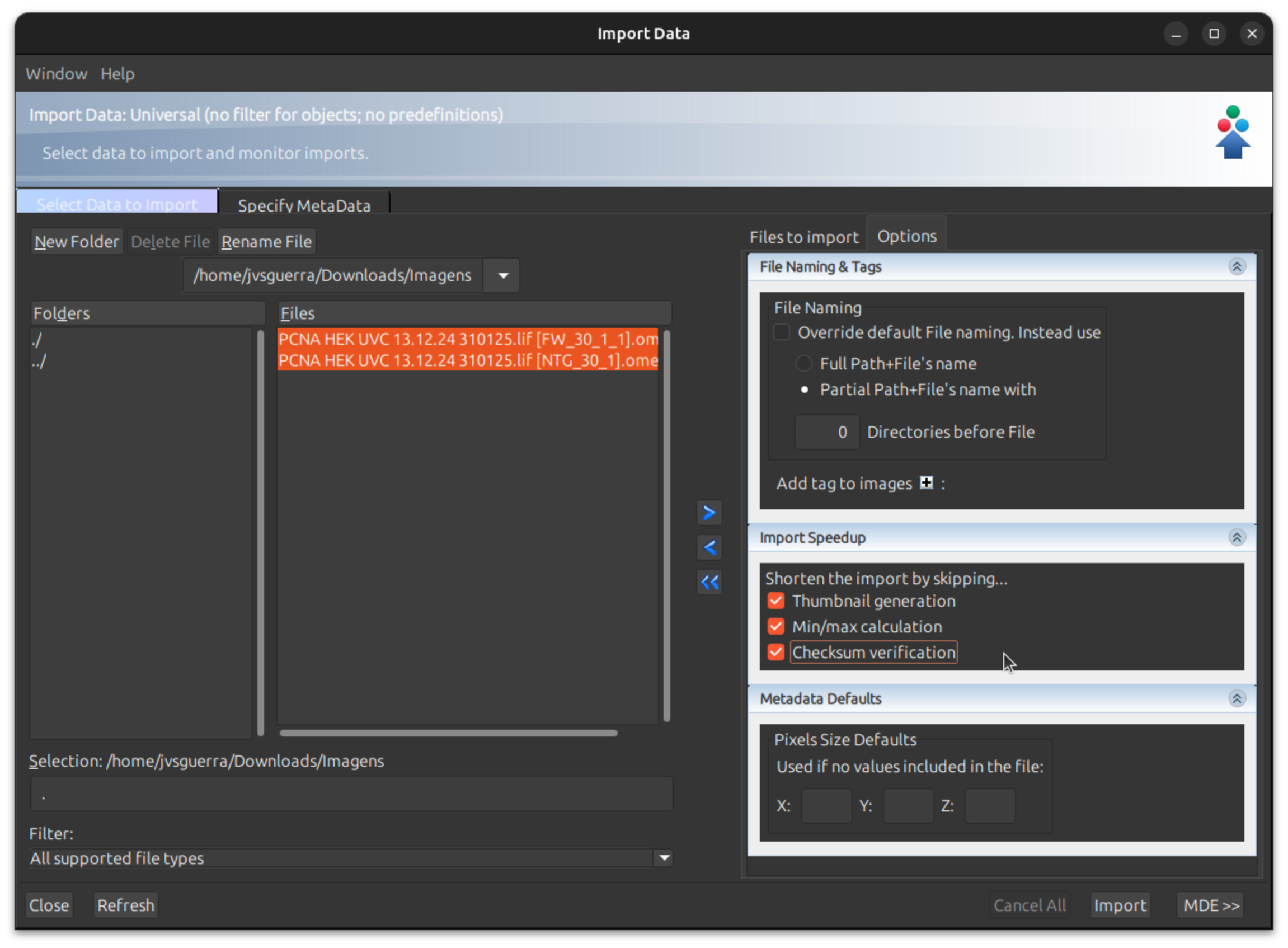
- Clique em “Import” para iniciar o processo de transferência das imagens para o OMERO.
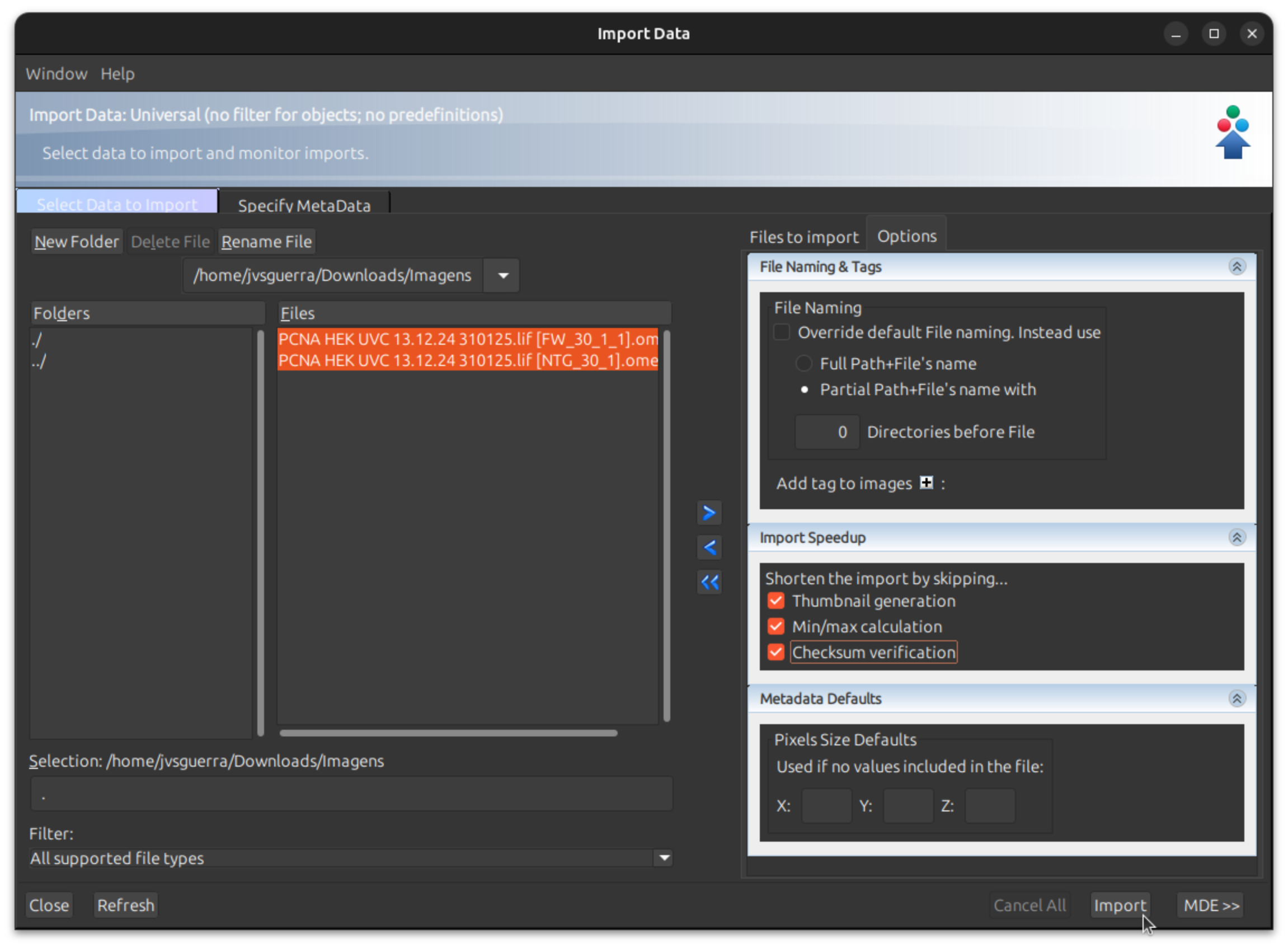
- Durante a transferência, uma janela de progresso será exibida, indicando os arquivos que estão sendo transferidos para o OMERO.
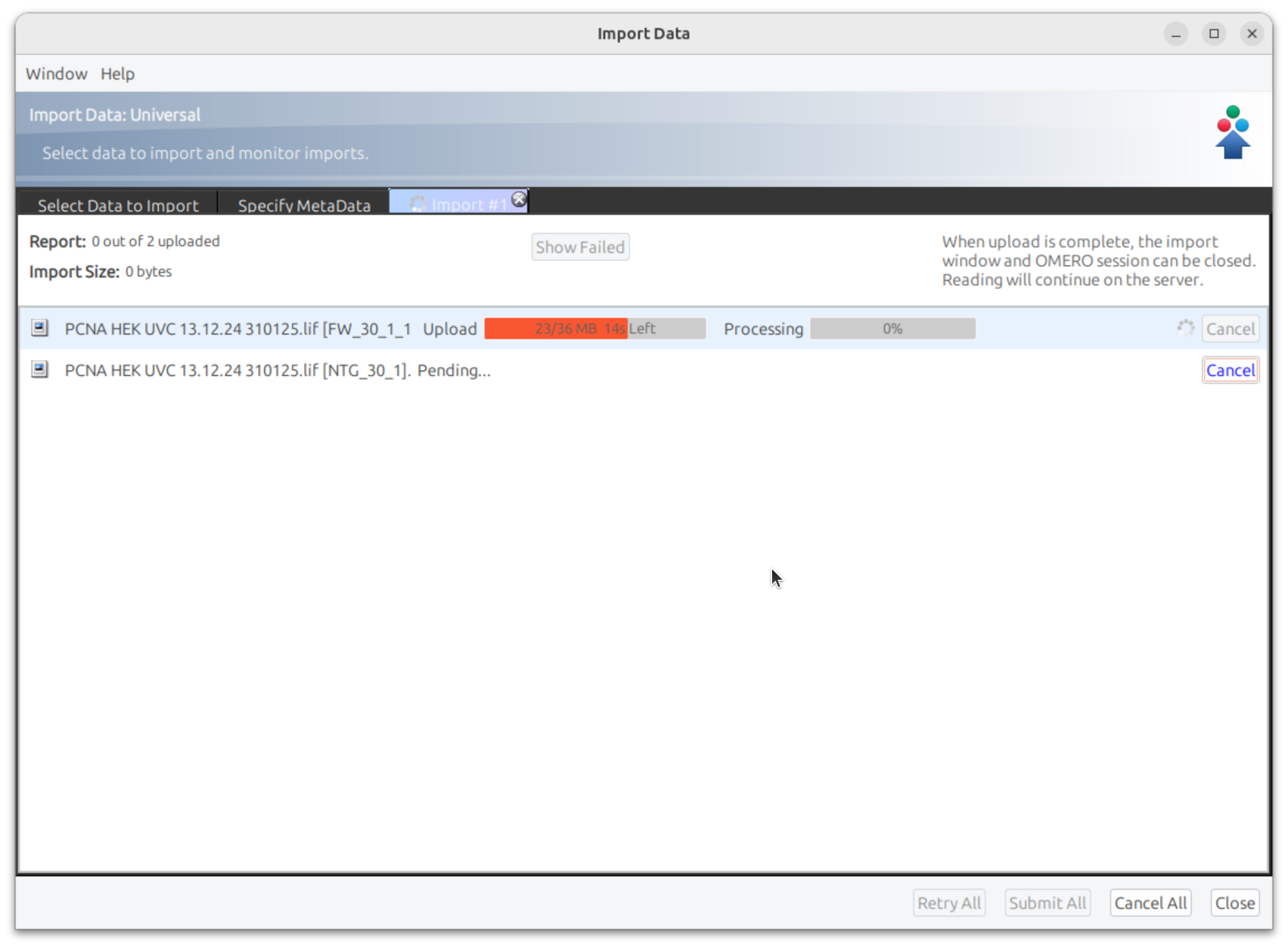
- Se a transferência for bem-sucedida, uma marcação verde (✔️) será exibido ao lado da aba ou no canto superior direito da tela inicial.
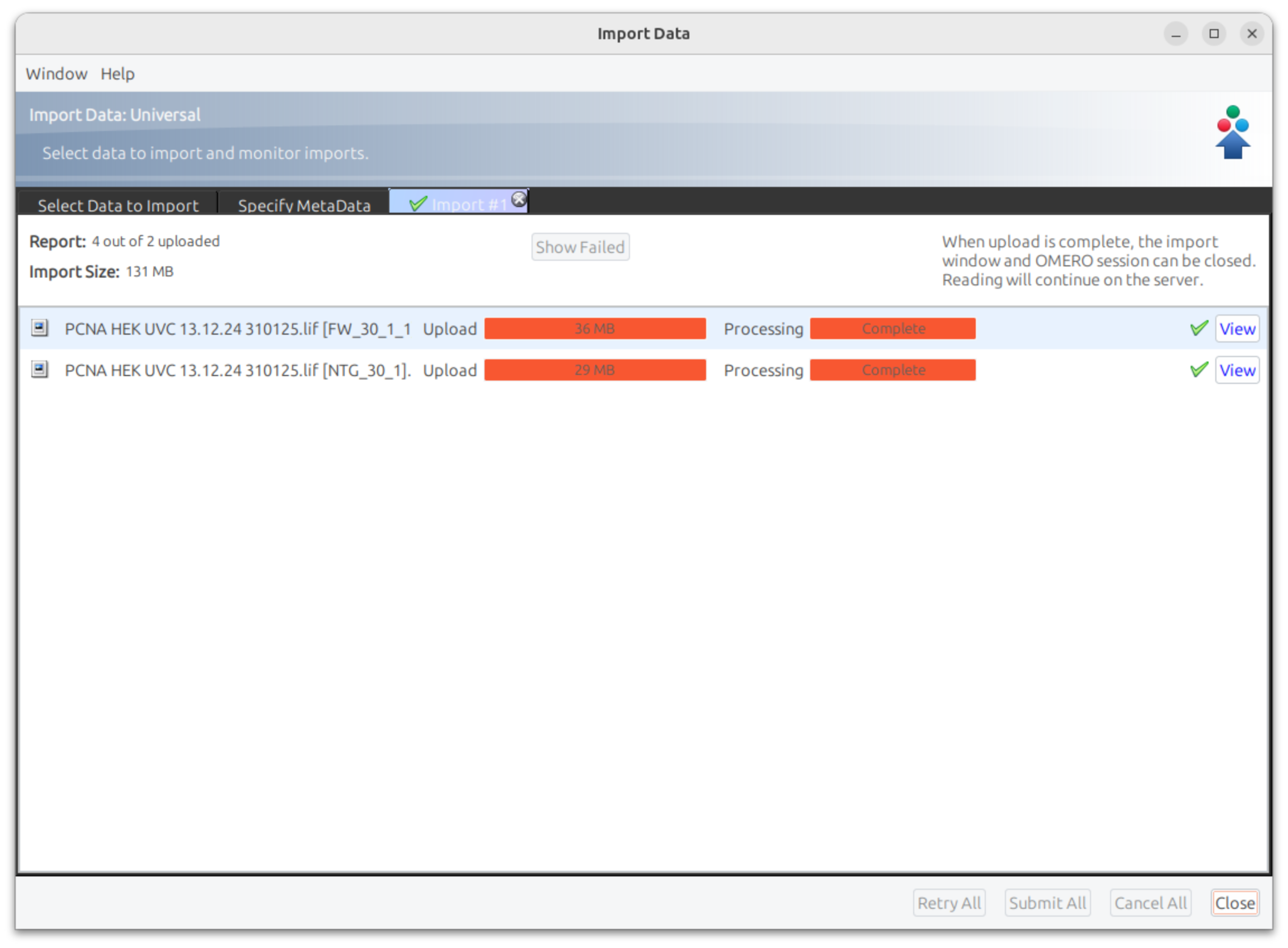
- Após a conclusão da transferência, as imagens estarão disponíveis no menu de navegação no projeto definido no momento da importação.
Caso não apareçam imediatamente, clique no ícone de “Refresh” (🔄) para atualizar as placas importadas pelo OMERO.insight.
Anotando imagens via OMERO.web
No OMERO.web, é possível executar scripts para realizar diversas tarefas, como gestão de anotação e exportação de dados. Para acessar o menu de scripts, clique no ícone de engrenagens no canto superior direito da interface, ao lado da barra de busca.
Os scripts estão organizados por categorias, sendo que os scripts voltados à manipulação de metadados estão localizados em annotation_scripts.
Tip
Selecione o objeto alvo (
Project,Dataset,Screen,Plate,Well,Image) na árvore de dados antes de abrir o script. Isso fará com que os camposData TypeeIDsejam preenchidos automaticamente.
Por que anotar as imagens?
A anotação rigorosa é essencial para a rastreabilidade científica. Metadados estruturados (Tags, pares Key-Value, comentários e anexos) são fundamentais para fluxos de análise automatizados e para a criação de conjuntos de dados alinhados aos princípios FAIR (Findable, Accessible, Interoperable and Reusable).
Expand Metadata: copiar anotações para objetos filhos
O script annotation_scripts > Expand Metadata automatiza a cópia de anotações de um objeto para seus “filhos” na hierarquia (ex: de um Projeto para todos os seus Datasets e Imagens).
Por exemplo, em invés de anotar manualmente a tag Tratamento em centenas de imagens, é possível anotar apenas o Screen de origem e utilizar o script para replicar a tag em todos os objetos (Plate, Well, Image) nele contido.
Parâmetros:
Data Type: O tipo de objeto selecionado como origem (Project,Dataset,Screen,Plate).ID: Identificador(es) do objeto. Para múltiplos alvos, separe os IDs por vírgula.Annotation Type: Tipo de metadado a ser copiado (Tags,Key-Value,File,CommentouAll).Source Level: Define em qual nível hierárquico o script deve buscar as anotações originais.
No exemplo abaixo, as tags de um objeto do tipo Screen (ID 1953) serão copiadas para seus objetos vinculados (Plate, Well e Image).
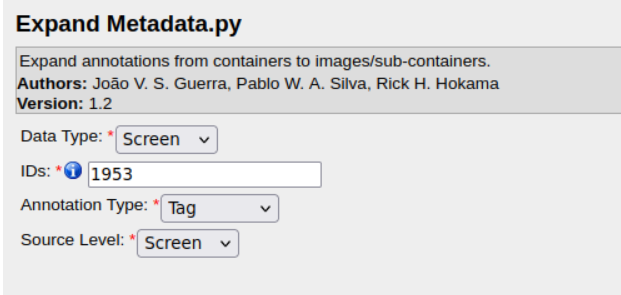
No exemplo seguinte, as tags dos Wells pertencentes ao Plate com ID 14220 seriam copiadas para os Images correspondentes aos Wells do Plate selecionado.
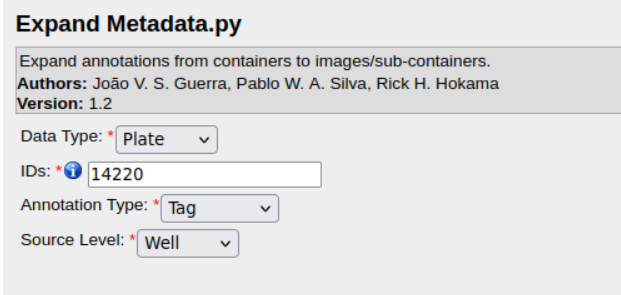
Clean Metadata: remover anotações
O script annotation_scripts > Clean Metadata permite a exclusão rápida e controlada de anotações, facilitando a correção de erros ou a padronização de grandes volumes de dados. Além de limpar os metadados de um objeto alvo, é possível deletar também as anotações de todos os objetos vinculados.
Parâmetros:
Data Type: O tipo de objeto selecionado como origem (Project,Dataset,Screen,Plate).ID: Identificador(es) do objeto. Para múltiplos alvos, separe os IDs por vírgula.Annotation Type: Tipo de metadado a ser removido (Tags,Key-Value,File,CommentouAll).Include Children: Se marcado, a remoção será aplicada também a todos os objetos vinculados ao alvo selecionado.
No exemplo abaixo, as tags de um objeto do tipo Screen (ID 1953) seriam removidas, sem afetar seus objetos vinculados (Plates, Wells e Images).
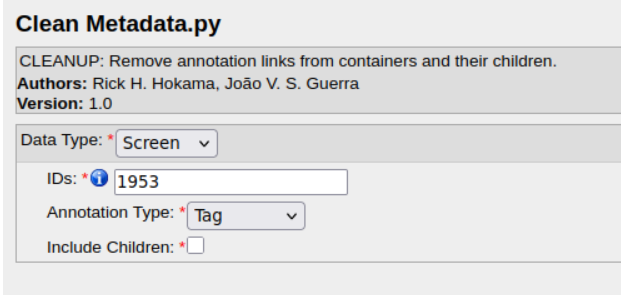
No exemplo seguinte, todas as anotações do Plate com ID 14220 seriam removidas. Como a opção de Include Children está ativada, as anotações dos Wells e Images associados também seriam excluídas.
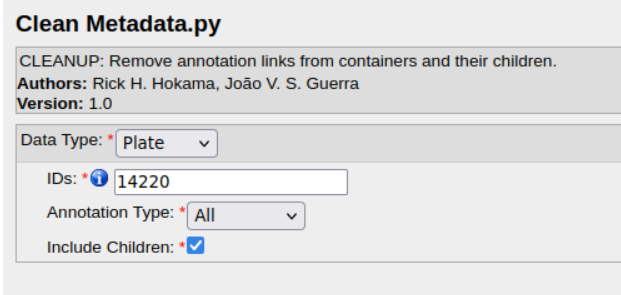
Boas práticas de anotação (FAIR no OMERO)
Para garantir que seus dados sejam FAIR (Findable, Accessible, Interoperable and Reusable), siga estas recomendações ao anotar imagens no OMERO:
- Seja consistente: utilize sempre os mesmos nomes para tags e pares key-value ao longo do projeto.
- Use vocabulários controlados: prefira termos padronizados (ex.: nomes de linhagens celulares, compostos, condições experimentais).
- Inclua metadados essenciais: anote informações críticas como linhagem celular, tratamento, tempo de exposição, tipo de microscopia, etc.
- Evite duplicações: antes de criar novas tags, verifique se elas já existem no OMERO utilizando a barra de busca.
Baixando imagens via OMERO.downloader
eLab
Esta seção apresenta o caderno eletrônico de laboratório eLab, que está disponível em https://elab.cnpem.br.
O eLab é um caderno eletrônico de laboratório (ELN) de código aberto, amplamente utilizado na comunidade científica para o registro e organização de dados experimentais. Ele permite que os pesquisadores documentem seus experimentos, compartilhem informações e colaborem de forma eficiente.
Para solicitar suporte ou ajuda com o eLab, registre um chamado na [LNBio] Suporte EDB do Jira em eLab: Suporte ao usuário.
Acesso pelo navegador 
Para acessar o eLab pelo navegador, abra seu navegador e acesse:
https://elab.cnpem.br
Na tela de login, use seu e-mail (p. ex., marie.curie@lnbio.cnpem.br) e senha institucional.
Suporte ao usuário
O suporte aos usuários do HPCC Marvin é realizado pela [LNBio] Suporte EDB do Jira.
Para solicitar ajuda com problemas técnicos, dúvidas sobre o sistema ou pedidos de recursos adicionais, registre um chamado na opção HPCC Marvin: Suporte ao usuário.
Para instalar ou atualizar um aplicativo ou programa, você deve registrar um chamado na opção HPCC Marvin: Programas e Aplicativos.
Ao abrir um chamado, por favor, forneça sempre informações relevantes, como o nome do usuário (marie.curie), o ID do job, a hora e a data em que ocorreu o problema e quaisquer mensagens de erro que você tenha recebido. Isso ajudará o suporte a identificar e resolver o problema com maior rapidez e eficiência.
Relatórios periódicos de uso do Marvin
Os relatórios periódicos de uso do Marvin são gerados anualmente, incluindo informações sobre o uso do sistema, como número de usuários, tempo de processamento, e tipos de trabalhos executados. Esses relatórios são úteis para entender a utilização do sistema e planejar melhorias.
Os relatórios anuais estão disponíveis no seguinte link: https://cnpem.github.io/marvin-reports/.
| Data de Publicação | Referente ao Ano |
|---|---|
| 2026-01-29 | 2025 |
| 2025-05-13 | 2024 |
| 2024-01-10 | 2023 |
Assistente de IA
Para auxiliar os usuários na utilização do ambiente de computação de alto desempenho (HPC), disponibilizamos dois agentes de IA personalizados. Eles estão configurados com as diretrizes deste manual para sanar dúvidas sobre o uso do HPCC Marvin.
-
Marvin (Gemini): Baseado no modelo Gemini do Google. Acessível a todos os usuários.
-
Marvin (Copilot): Baseado no modelo Microsoft Copilot. Acesso exclusivo para usuários com conta institucional do CNPEM.
Canais de Acesso
Treinamentos
- Treinamento em Biologia Computacional (2025)
- I Ciclo de Treinamentos em Microscopia de Luz (2026)